
Abstract
Metastasis remains the leading cause of cancer-related death. In 1889, Stephen Paget originally proposed the theory “seed-and-soil.” Both cancer cell-intrinsic properties (“seed”) and fertile microenvironment (“soil”) are essential for metastasis formation. To date, accumulating evidences supported the theory using mouse models. The formation of a premetastatic niche has been widely accepted as an accel for metastasis. Similar to tumor microenvironment, various types of cells, such as immune cells, endothelial cells, and fibroblasts are involved in premetastatic niche formation. We have discovered that primary tumors hijack Toll-like receptor 4 (TLR4) signaling to establish a premetastatic niche in the lung by utilizing the endogenous ligands. In this review, we discuss the mechanisms that underlie inflammation-associated premetastatic niche formation upon metastasis, focusing especially on myeloid cells and macrophages as the cells executing and mediating complicated processes.
Similar content being viewed by others


Background
Metastasis consists of a continuous and multi-step biological process, in which a small population of cancer cells with highly invasive and metastatic potential secede from their primary tumors, induce degradation of the extracellular matrix (ECM), intravasate into the blood or lymphatic vessels, and extravasate to and colonize to distant organs. The theory of “seed and soil” proposed by Paget [1] has been widely accepted. To date, secreted cytokines, such as vascular endothelial growth factor (VEGF), stromal cell-derived factor 1(SDF-1), transforming growth factor b (TGF-b), and tumor necrosis factor (TNF), which form premetastatic niches, induce mobilization of bone marrow-derived cells. The recruitment of vascular endothelial growth factor receptor 1 (VEGFR1)+ hematopoietic bone marrow progenitors, CD11b+ myeloid cells and suppressive immune cells, initiates the pre-metastatic niches and thereby promotes metastasis [2]. We previously identified S100A8 which was upregulated in the lungs of tumor-bearing mice [3]. S100A8 is one of the known endogenous ligands of TLR4 [4].
Toll-like receptors (TLRs), the best-characterized family of pattern recognition receptors, were originally characterized for their ability to respond to exogenous pathogen-associated molecular patterns (PAMPs) that include bacterial lipopolysaccharide (LPS), bacterial diacylated and triacylated lipopeptides, bacterial flagellin, bacterial and viral unmethylated CpG-containing DNA motifs, and viral single- and double-stranded RNA [5]. Among TLRs, TLR4 activation via LPS is essential for the host defense against gram-negative bacteria. In addition to PAMPs, TLR4 also recognizes danger-associated molecular patterns (DAMPs). Heat-shock protein (Hsp) 60 [6], Hsp70 [7], high-mobility group box 1 (HMGB1) [8], S100A8/S100A9 [4], serum amyloid A3 (SAA3) [9, 10], Fetuin-A [11], defensin β [12], fibrinogen [13], and fibronectin [14], as well as polysaccharides such as hyaluronan [15], heparan sulfate [16], biglycan [17], and decorin [18], appear to be endogenous ligands of TLR4. The ligand-induced dimerization of TLRs triggers the recruitment of adaptor proteins to intracellular TIR (Toll/interleukin-1 receptor) domains to initiate signaling. Signaling cascades via the TIR domains are mediated by specific adaptor molecules, including MyD88, MAL (also known as TIRAP), TRIF, and TRAM. TLR4 associates with co-receptor MD-2 and LPS binding to MD-2 induces the dimerization of TLR4 [19]. Intriguingly, accumulating evidence suggest that DAMP-mediated signals can promote metastasis. Initially, we thought that S100A8 would mainly play a role in premetastatic niche formation, but it has been reported that various types of cancer cells also by themselves express TLR4 at the cell surface. To date, S100A8 expression, or overexpression of TLR4, was observed in various cancers.
Myeloid derived suppressor cells (MDSCs) in metastasis
MDSCs are immature myeloid cells derived from bone marrow, recruit to primary tumor sites and distant organs, which contribute to various immune responses. The recruitment of MDSCs is a crucial step for premetastatic niche formation [20,21,22]. The ability of immunosuppression is one of the major characteristics of MDSCs. In mice, MDSCs can be divided at least into two populations, namely granulocytic/polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). In addition, early MDSCs (eMDSC) have been identified in humans. Classical myeloid cell activation is mainly driven via TLR activation. Both M-MDSCs and PMN-MDSCs express S100A8/S100A9. PMN-MDSCs in the premetastatic niches may contribute to the escape of tumor cells by suppressing immune cells, inducing matrix remodeling, and promoting angiogenesis, which in turn promote colonization of tumor cells. In mice, M-MDSCs are defined as CD11b+Ly6G–Ly6Chigh, and PMN-MDSCs are defined as CD11b+Ly6G+Ly6Clow. It should be noted that these markers are only applicable for mice, but not for humans. In humans, M-MDSCs are originally defined as either CD14+HLA−DLlow or CD11b+CD14−CD33+CD15− cell populations, whereas PMN-MDSCs are defined as CD11b+CD14−CD15+ or CD11b+CD14−CD66+ cells.
Tumor associated macrophages (TAMs) and metastasis-associated macrophages (MAMs)
TAMs, one of the most abundant inflammatory stromal cells in the tumor microenvironment, exhibit predominantly M2-like pro-tumor phenotype rather than M1-like anti-tumor phenotype, and their importance in metastasis is well established [23]. The subpopulation of recruited MDSC from the bone marrow differentiates to TAMs. Similar to MDSC, TAMs can induce immunosuppressive cytokines. Blockade of the CSF1–CSF1R with anti-CSF1 antibodies or a CSF-1R inhibitor with chemotherapy enhanced the therapeutic efficacy, inhibited metastases, and increased the recruitment of CD8 T cells in tumors [24].
Moreover, metastasis-associated macrophages (MAMs) have been identified in murine experimental models as F4/80+ CD11b+CD11c−Ly6C− cells [25]. Of note, both TAMs and MAMs are originated from the bone marrow, but not residential macrophages. Lu et al. reported that TAM interacted with cancer stem cells, which would maintain cancer stemness [26]. These results indicate that the interaction in primary tumor sites contributes to maintain an invasive phenotype.
TLRs in premetastatic niche formation
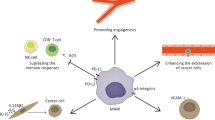
TLR2, TLR3, and TLR4 have been reported that TLR-mediated signaling involves premetastatic formation (Fig. 1). We focused on these three receptors and their ligands to discuss the mechanisms by referring to their findings.

TLRs involved in metastasis. TLR2 associates with TLR1 or TLR6. TLR4 recognizes LPS or endogenous ligands (i.e., biglycan, HMGB1, S100A8, S100A9). TLR3 is implicated in the recognition of viral dsRNA or exosomal RNA. TLR2, TLR3, or TLR4 are involved in cancer metastasis
TLR2
Kim et al. firstly reported that versican induces metastasis in a TLR2-dependent manner. Versican is one of the TLR2/6 endogenous ligands, which can activate TLR2 downstream signaling. The deficiency of TLR2 in host cells significantly reduced pulmonary metastasis [27]. Moreover, Gao et al. found that bone marrow-derived CD11b+Gr1+ myeloid progenitor cells were recruited to premetastatic lung niches in the PyMT spontaneous breast cancer model [28]. In these niches, a specific subpopulation of myeloid cells, CD11b+Ly6Chigh produced the ECM proteoglycan versican that stimulated mesenchymal-to-epithelial transition of metastatic tumor cells, which induced proliferation and hence the development of macrometastases [28].
TLR3
The secreted molecules by the primary tumor also contain exosomal RNAs. Liu et al. reported that TLR3 in alveolar type II (AT-II) epithelial lung cells by tumoral-exosome-derived non-coding snRNA-mediated TLR3 activation in alveolar type II epithelial lung cells contributes to neutrophil (possibly contains PMN-MDSCs) infiltration and lung metastasis [29]. TLR3-deficient mice show reduced pulmonary metastasis in the spontaneous metastatic mouse models. These results suggest that not only immune cells but also other host cells, in this case resident alveolar epithelial cells, can involve in premetastatic niche formation.
TLR4
We demonstrated that secretion of VEGF-A, TNFα, and TGF-β by primary tumors induces expression of S100A8/S100A9 in the lung, which causes a fertile microenvironment for metastasis (Fig. 2). S100A8 is known to be an endogenous ligand of TLR4 which can induce serum amyloid A (SAA) 3 in the premetastatic lung. Essentially, among all TLR series, only TLR4 can activate two downstream signalings upon activation by PAMPs. This resulted in the accumulation of CD11b+ myeloid cells and propagated a positive feedback loop for further chemoattractant secretion, all of which led to enhanced metastasis in the lung [3, 9]. In addition to the induction of S100A8 in the premetastatic lung, upregulation of S100A8 in the tumor microenvironment, which involves into the recruitment of myeloid cells in lungs from the bone marrow to primary tumors. Moreover, we further found that TLR4 knockdown cells grew slower than parental cells in vivo, whereas TLR4 knockdown cells grew similar to parental, or scramble shRNA-infected cells in 2D culture, indicating that tumor microenvironmental factors secreted by host immune cells are necessary to induce TLR4-mediated tumor growth [30]. To investigate whether TLR4 inhibition is sufficient to suppress tumor progression, Eritoran was used for further study. Eritoran is a structural analog of the lipid A from R sphaeroides (RsLA), competitively binds to TLR4/MD-2, which results in inhibition of LPS-induced inflammatory responses [31]. We found that Eritoran blocked S100A8-mediated TLR4 activation and reduced TAMs and CD11b+Ly6C++Ly6G− populations in tumor microenvironment. Conversely, the treatment with Eritoran in tumor-bearing mice significantly increased intratumor infiltrating CD8+ T cells compared with the tumor-bearing control mice. The suppression of T cell function by MDSCs more likely depends on Arginase-1 expression [30].

The recruitment of myeloid-derived cells is a crucial step in metastasis. In the early phase of the primary tumor, it is not ready for myeloid-derived cells (inflammatory monocytes, MDSCs) to recruit primary tumor sites and distant organs (in this case, the lungs; do not walk). Once tumors develop to a certain size and acquire invasiveness, the primary tumor microenvironment secretes cytokines/chemokines (i.e., VEGF, TNFα, and TGF-β) to produce S100A8 in the lungs. These chemokines, such as Ccl2, S100A8/S100A9, can send a signal to myeloid-derived cells in bone marrow as “walk”
Fukata et al. reported that mice with constitutive active TLR4 in epithelial cells were highly prone to colitis and colitis-associated colorectal tumor [32]. Stimulation of TLR4 signaling induces cyclooxygenase-2, which causes PGE2 production followed by upregulation of amphiregulin. In skin cancer, TLR4 also appears in keratinocytes and levels up during their progression to squamous cell carcinoma (SCC), whereas silencing of TLR4 led to the blockade of UV stress [33]. Moreover, Fleming et al. recently reported that tumor cell-derived extracellular vesicles convert normal myeloid cells into functional MDSCs by regulating the expression of PD-L1 via TLR4 signaling [34]. TLR4-induced TGF-β expression has also been associated with the transformation of fibroblast into cancer-associated fibroblast (CAF) in the tumor microenvironment, facilitating cancer cell proliferation and tumor growth [35, 36]. Therefore, targeting the premetastatic niche-promoting molecular components to prevent metastasis may be an attractive approach for cancer therapeutics.
S100A8/S100A9
S100A8 belong to the S100s family, exclusively expressed in vertebrates and characterized by two calcium-binding EF-hand motifs: a carboxyl-terminal EF-hand (high affinity to calcium) and an N-terminal EF-hand (low affinity to calcium) and connected by a central hinge. Calcium-binding within the EF-hand motif occurs in response to increased intracellular calcium concentrations and causes conformational changes that expose a wide hydrophobic cleft that interacts with target proteins. Like most S100 proteins, S100A8 can form as a heterodimer/heterotetramer with S100A9 and as a homodimer or monomer [37]. Vogl et al. originally reported that S100A8-mediated cytokine induction was mediated by TLR4 [4]. Moreover, we found that carboxyl terminal region of mouse S100A8 has a high affinity to TLR4/MD-2 and that S100A8 induced inflammatory cytokines/chemokines in a TLR4-dependent manner [30]. Although it has been reported that the S100A8/S100A9 heterodimer complex promotes colon tumor progression via the RAGE-dependent pathway [38], S100A8-mediated TLR4 activation in host cells could play a role in inflammatory cytokine/chemokine production.
Biglycan
Secreted proteoglycans such as biglycan, decorin, and versican can act as endogenous ligands of TLRs. Biglycan, a member of the family of small leucine-rich proteoglycan, is a ubiquitous ECM component. Biglycan acts as an endogenous ligand of both TLR2 and TLR4. Schaefer et al. reported that biglycan can bind to both TLR2 and TLR4 directly, but not TLR3, 5, 7, or 8 [17]. Recently, Maishi et al. reported that biglycan secreted from tumor endothelial cells induced tumor cell migration through NFκB and ERK2 activation via TLR2/4, which resulted in tumor metastasis [39]. In breast, lung, and colorectal cancers, high expression of biglycan correlated with poor prognosis [39]. In addition, Cong et al. found that biglycan-deficient mice showed vascular normalization in the tumor microenvironment, which enhances the efficacy of chemotherapy [40].
HMGB1
High-mobility group box 1 (HMGB1), as a member of the HMG family, predominantly locates in the nucleus as a DNA chaperone, which also translocates to cytoplasm, as well as the outside of the cell as a DAMP molecule. Because HMGB1 knockout mice die shortly after birth, HMGB1 is essential for development. HMGB1 is ubiquitous in mammals and generally abundant. HMGB1 is secreted from dead cells or activated immune cells, enterocytes, hepatocytes, and possibly several other cell types during infection, injury, or severe stress [41].
HMGB1 comprises two DNA-binding domains (so-called Box A and Box B) and acidic C-terminal tail. Since HMGB1 lacks a signal sequence, secretion occurs with the fusion of vesicles with the plasma membrane after an inflammatory signal. According to this, NLRP3 inflammasome might have an important role in the secretion of HMGB1 [42]. HMGB1 interacts not only with TLR4, but also with TLR2, and the receptor for the advanced glycation end product (RAGE). Yang et al. reported that HMGB1 failed to induce inflammatory cytokines in TLR4-deficient macrophages. It is still unclear how HMGB1 chooses a specific receptor, but it has been reported that TLR4 but not RAGE is required for cytokine release [8] and that RAGE is involved in cell migration [43].
Among endogenous TLR4 ligands, HMGB1 is studied in detail by using HMGB1 protein of endotoxin-free grade or synthetic peptide (B Box (89–108)). Endotoxin-free grade of HMGB1 or B Box peptide binds to TLR4/MD-2 in a concentration-dependent manner, whereas Cys106Ala peptide failed to bind to TLR4/MD-2. Therefore, Cys106 residue of HMGB1 is required for the binding to TLR4/MD-2 and activation of inflammatory cytokine production [8]. In addition to the importance of Cys106, TLR4 activation by HMGB1 requires a concomitant disulfide Cys23-Cys45 linkage [44]. Accordingly, the redox state is important for HMGB1-dependent TLR4 activation. More recently, Yang et al. developed tetramer FSSE to inhibit HMGB1-MD-2 interaction, while sparing PAMP, like LPS, signaling through TLR4 [45].
The blockage of HMGB1 can be applied as a therapeutic purpose against cancer disease? Nadatani et al. reported that administration of non-steroidal anti-inflammatory drugs induced small intestinal damage through a TLR4-dependent manner, and was associated with an increase of HMGB1 in both intestine and serum. The blockage of HMGB1 with neutralizing antibodies prevented both tissue damage and inflammatory cytokine production [46]. It has been reported that HMGB1 increased MDSC-mediated IL-10 production [47]. These findings suggest that a strategy of HMGB1 blockage is one of the useful clinical approaches against inflammatory-related diseases.
S100A8-TLR4/MD-2 signaling can be a therapeutic target against cancer progression/metastasis?
As mentioned above, we found that Eritoran, a TLR4 inhibitor, inhibits tumor growth by immunomodulation and vascular normalization. Administration of anti-S100A8 neutralizing antibodies also suppresses tumor progression by the suppression of MDSC recruitments [30]. Of note, the neutralizing antibodies inhibit not only lung metastasis, but also liver metastasis (Deguchi, unpublished data). Initially, we thought that S100A8 mainly would play a role in premetastatic niche formation, but it has been reported that various types of cancer cells also by themselves express TLR4 at the cell surface.
Therefore, S100A8-TLR4/MD-2 may play crucial roles in both host cells and primary tumors. Accordingly, the S100A8-TLR4/MD-2 axis could be a novel therapeutic target against cancer progression and metastasis by immunomodulation and vascular normalization.
Existence of metastasis-suppressive cell population in premetastatic phase
We have learned that premetastatic niche formation contributes to metastasis; however, it is likely that some suppressive effects would also exist to maintain homeostasis during the premetastatic phase. Moreover, until recently, the mechanisms of metastasis suppression are poorly understood. The bone marrow-derived Gr-1+ myeloid cells which express thrombospondin-1 suppress metastasis, and primary tumor-derived prosaposin play a key role in thrombospondin-1 induction in the Gr-1+ cells [48]. Of note, the expression of prosaposin in prostate cancer was positively correlated with increased overall survival. More recently, we investigated the suppressive role of metastasis during the premetastatic phase and identified the population of B220+CD11b+NK1.1+ cells as a pulmonary metastasis suppressor. These results suggested that hepato-entrained B220+CD11b+NK1.1+ cells suppressed lung metastasis [49]. These findings would provide a novel clue for therapeutic targets against metastatic tumors.
Conclusions
We now realize that many types of immune cells complexly contribute to premetastatic niche formation for immune escape. Chemoresistance or recurrence are major causes of cancer-related death. Elucidation of the underlying mechanism(s) can contribute to establishing suitable therapeutic targets. Of note, metastatic tumors tend to disseminate to specific distant organs. For example, aggressive colon cancers typically metastasize to the liver. We found that S100A8 plays an important role in the premetastatic niche formation, but not only to the lungs specifically. Our results suggested that S100A8 universally induce the cell mobilization of MDSCs and other inflammatory monocytes from the bone marrow. As mentioned above, S100A8 is one of the major inflammatory cytokines in acute and chronic inflammation. Intriguingly, S100A8 expression is known to be upregulated in severe COVID-19 patients [50, 51]. Therefore, in addition to cancer disease, it is likely that S100A8/TLR4/MD-2 axis can be a promising therapeutic target against other inflammation-associated diseases.
Availability of data and materials
Not applicable.
Abbreviations
- TRIF:
-
TIR domain-containing adaptor protein-inducing interferon β
- TRAM:
-
TRIF-related adaptor molecule
- MyD88:
-
Myeloid differentiation factor 88
- MAL:
-
MyD88 adaptor-like
- CSF1:
-
Colony-stimulating factor 1
- CSF1R:
-
Colony-stimulating factor 1 receptor
- NLRP3:
-
NLR family pyrin domain containing 3
- PGE2 :
-
Prostaglandin E2
References
Paget S. The distribution of secondary growths in cancer of the breast. Lancet. 1889;133:571–3.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the premetastatic niche. Nature. 2005;438:820–7.
Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. 2006;8:1369–75.
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–9.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84.
Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, et al. Endocytosed HSP60s use Toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–9.
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–34.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107:11942–7.
Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–55.
Deguchi A, Tomita T, Omori T, Komatsu A, Ohto U, Takahashi S, et al. Serum amyloid A3 binds MD-2 to activate p38 and NF-kappaB pathways in a MyD88-dependent manner. J Immunol. 2013;191:1856–64.
Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18:1279–85.
Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–9.
Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through Toll-like receptor 4. J Immunol. 2001;167:2887–94.
Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–33.
Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9.
Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–9.
Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–33.
Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011;4:ra75.
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5.
Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol. 2009;85:996–1004.
Kitamura T, Qian BZ, Pollard J. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86.
Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021. https://doi.org/10.1038/s41577-020-00490-y.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61.
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67.
Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–59.
Lu H, Clauser KR, Tam WL, Fröse J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16(11):1105–17.
Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–6.
Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 2012;72:1384–94.
Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell. 2016;30:243–56.
Deguchi A, Tomita T, Ohto U, Takemura K, Kitao A, Akashi-Takamura S, et al. Eritoran inhibits S100A8-mediated TLR4/MD-2 activation and tumor growth by changing the immune microenvironment. Oncogene. 2016;35:1445–56.
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–17.
Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–81.
Blohm-Mangone K, Burkett NB, Tahsin S, Myrdal PB, Aodah A, Ho B, et al. Pharmacological TLR4 antagonism using topical resatorvid blocks solar UV-induced skin tumorigenesis in SKH-1 mice. Cancer Prev Res. 2018;11:265–78.
Fleming V, Hu X, Weller C, Weber R, Groth C, Riester Z, et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating PD-L1 via TLR4 signaling. Cancer Res. 2019;79:4715–28.
Bhowmick NA. TGF-ß signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51.
Seki E, De Minicis S, Österreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat Med. 2007;13:1324–32.
Kumar RK, Yang Z, Bilson S, Thliveris S, Cooke BE, Geczy CL. Dimeric S100A8 in human neutrophils is diminished after phagocytosis. J Leukoc Biol. 2001;70:59–64.
Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011;9:133–48.
Maishi N, Ohba Y, Akiyama K, Ohga N, Hamada J, Nagao-Kitamoto H, et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci Rep. 2016;6:28039.
Cong L, Maishi N, Annan DA, Young MF, Morimoto H, Morimoto M, et al. Inhibition of stromal biglycan promotes normalization of the tumor microenvironment and enhances chemotherapeutic efficacy. Breast Cancer Res. 2021;23:51. https://doi.org/10.1186/s13058-021-01423-w.
Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4-dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–23.
Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–92.
Penzo M, Molteni R, Suda T, Samaniego S, Raucci A, Habiel DM, et al. Inhibitor of NF-kB kinases a and b are both essential for high mobility group box 1-mediated chemotaxis. J Immunol. 2010;184:4497–509.
Yang H, Lündback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. 2012;18:250–9.
Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14.
Nadatani Y, Watanabe T, Tanigawa T, Machida H, Ozaki H, Yamagami H, et al. High mobility group box 1 promotes small intestinal damage induced by nonsteroidal anti-inflammatory drugs through Toll-like receptor 4. Am J Pathol. 2012;181:98–110.
Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res. 2014;74:5723–33.
Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov. 2013;3:578–89.
Hiratsuka S, Tomita T, Mishima T, Matsunaga Y, Omori T, Ishibashi S, et al. Hepato-entrained B220+CD11c+NK1.1+ cells regulate pre-metastatic niche formation in the lung. EMBO Mol Med. 2018;10:e8643.
Silvin A, Chapuis N, Dunsmore G, Goubet AG, Dubuisson A, Derosa L, et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182:1401–1418.e18.
Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27:883–890.e2.
Acknowledgements
The authors thank all our lab members for the helpful discussion.
Funding
This work is supported in part by Grants-in-Aid for Scientific Research by the Japanese Society for the Promotion of Science (JP18K07245 and JP21K07157 to AD, JP19K03506 to YM), the Takeda Science Foundation (to AD), and the Astellas Foundation for Research on Metabolic Disorders (to AD).
Author information
Authors and Affiliations
Contributions
AD and YM wrote the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deguchi, A., Maru, Y. Inflammation-associated premetastatic niche formation. Inflamm Regener 42, 22 (2022). https://doi.org/10.1186/s41232-022-00208-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41232-022-00208-8