
Abstract
Background
Geographical patterns and degrees of genetic divergence among populations differ between species, reflecting relative potentials for speciation or cladogenesis and differing capacities for environmental adaptation. Identification of factors that contribute to genetic divergence among populations is important to the understanding of why some species exhibit greater interpopulation genetic divergence. In this study, we calculated the mean pairwise genetic distances among populations as species’ average genetic divergence by a phylogeny using nuclear and mitochondrial genes of 303 individuals from 33 Cuban Anolis species and estimated species ages by another phylogeny using nuclear and mitochondrial genes of 51 Cuban and 47 non-Cuban Anolis species. We identified factors that influence species’ differences in genetic divergence among 26 species of Anolis lizards from Cuba. Species ages, environmental heterogeneity within species ranges, and ecomorph types were considered as factors affecting average genetic divergences among populations.
Results
The phylogenies presented in this study provide the most comprehensive sampling of Cuban Anolis species to date. The phylogeny showed more conservative evolution of Anolis ecomorphs within Cuba and identified twig anoles as a monophyletic group. Subsequent Phylogenetic Generalized Least Squares (PGLS) analyses showed that species age was positively correlated with species’ average genetic divergence among populations.
Conclusion
Although previous studies have focused on factors affecting genetic divergence within species, the present study showed for the first time that species differences in genetic divergence could be largely affected by species age.
Similar content being viewed by others


Background
Genetic divergence among populations contributes to ecological divergence and subsequent speciation [1]. Additionally, greater genetic divergence with limited gene flows among populations reportedly promotes local adaptation and acquisition of potential to inhabit various environments within the range [2]. Thus, the identification of factors that contribute to genetic divergence among populations is useful to gaining a better understanding of why some species have greater interpopulation genetic divergence than others. Previous studies have mainly focused on factors that affect intraspecific differences in the genetic divergence among populations. For instance, it has been reported that geographically distant populations are likely to experience isolation owing to the distance [3]; thus, geographical barriers and distances may limit gene flow. Furthermore, genetic divergence among populations from different environments may be larger, owing to selection against individuals that move between the environments [4]. Hence, in addition to geographical isolation, isolation by environment is a major determinant of intraspecific genetic differentiation between populations, and the relative contributions of these factors and population histories have been investigated in several studies [5, 6].
However, average genetic divergence among populations also differs between species, and factors that affect interspecific differences in these genetic divergences among populations likely differ from those that affect intraspecific differences. Among potential predictors of average genetic divergence among populations, differences in species’ dispersal abilities or tendencies, which could be affected by several factors, influence gene flow and subsequently genetic divergence among populations. For instance, morphologically different species may differ in their dispersal ability [7, 8]. In addition, the effect of isolation based on environment might differ among species because individual dispersal tendencies with respect to preference of particular environments may have evolved depending on selective pressure and past environments experienced by the species.
Phylogenetic history [9] and species age [10] are crucial determinants of genetic divergence patterns and are responsible for current geographical species range. In particular, geographical distances over which gene flows occur increase with species ranges, and greater genetic differentiation has been shown among populations of various older species [9, 11,12,13,14]. However, few studies demonstrate the effects of species age on genetic divergence among populations within a species, and although Fujisawa, Vogler and Barraclough [15] correlated species occupancy (range size) with genetic variations in mitochondrial DNA sequences within species, correlations with species age were not identified.
Anolis lizards are among the most diverse vertebrate genera, and about 120 or more species have been identified in the Greater Antilles countries of Cuba, Hispaniola, Jamaica, and Puerto Rico [16]. Anolis lizards display a wide range of morphological and behavioral adaptations that are closely related to their most frequently used microhabitats. Hence, sets of species that share morphological, ecological, and behavioral traits are grouped into different ecological types, and are referred to as ecomorphs [17]. Cuba is the largest island in the Caribbean and has the highest diversity of Anolis lizards, with 64 known species [18]. Although classifications of ecomorphs vary between studies, Cuban ecomorphs have been defined as crown-giant, trunk-crown, trunk-ground, twig, and grass-bush. Although these ecomorphs have independently evolved on multiple Caribbean islands, some species show unique and independent evolution on islands that lacks ecomorphological classifications. Such anoles are considered to be unique and can be found within different clades among the Cuban Anolis [19].
Previous studies have indicated deep interpopulation genetic divergence in several Greater Antillean Anolis species [20,21,22,23,24,25,26], and deep mitochondrial divergence has also been demonstrated in species from the Lesser Antilles [27,28,29,30,31]. These studies show comparatively high levels of genetic divergence among populations of some species, leading to large differences in interpopulation genetic divergence among Anolis species. In a recent study, Wang, Glor and Losos [6] quantified relative influences of ecological (local environmental conditions) and geographical (distances between locations) factors on spatial genetic divergence in Anolis species from the Greater Antilles islands from Cuba, Hispaniola, Jamaica, and Puerto Rico. They showed that, although both geographical and ecological isolation significantly influence genetic divergence, the geographical isolation was substantially more predictive, suggesting that non-ecological factors play dominant roles in the evolution of spatial genetic divergences [6]. Wang et al. [6] also demonstrated that the effect of geographical distance on genetic divergence among populations was greater in some Anolis species than that in others; however, their study did not explore why some species have greater interpopulation genetic divergence than others.
In the present study, we identified determinates of interspecific (or interclade) differences in genetic divergence among populations of Cuban Anolis species. Specifically, we considered species age, environmental heterogeneity within species ranges, and ecomorph type as putative factors and tested the following hypotheses: (1) Older species are more likely to show greater genetic differentiation due to the greater time available for divergence. The effect of age on divergence can be detectable if the homogenizing effects of gene flows are not significantly large; (2) Species inhabiting more heterogeneous environments have evolved to adapt to different environments and thus have larger genetic divergence among populations than species inhabiting homogeneous environments; (3) Species that belong to different ecomorphs live in different microhabitats and show different moving abilities, which correlate with interspecific differences in genetic divergence.
Average genetic divergence is positively correlated with the average geographical distance among populations and this, in turn, is positively correlated with the size of the species’ range, which sometimes depends on the sampling locations between which the geographical distances are measured. Therefore, the average geographical distance can be used as a covariate of genetic distance and the above three factors could be examined by considering the effect of the average geographical distance for each species.
To test the hypotheses stated above, we used independent monophyletic clades as units of comparison. The members of populations within the clades potentially interbreed, so we examined factors affecting different degree of gene flows among populations within the clades. In addition, studied clades need to be independent and monophyletic so that the effects of population branching and separation from common ancestors can be assessed without gene flows from other clades. Using these units, we are able to evaluate whether genetic divergence within the clades was affected by emergent characteristics of the clade (e.g., environmental heterogeneity within species range, species age, etc.) or traits shared with all the populations of the species (e.g., morphology). In this study, in principle, we used species, but as shown below, some species form paraphyletic, not independent monophyletic clades. In such cases, we used independent monophyletic clades within the species as comparative units.
Herein, we focused on Anolis lizards in Cuba, since Cuba has the highest diversity of Anolis lizards among the Caribbean islands. We reconstructed a phylogeny for 33 Cuban Anolis species using nuclear and mitochondrial genes, and estimated pairwise genetic distances among populations for 26 of these. We also estimated divergence times using a reconstructed phylogenetic tree of 55 Cuban and 47 non-Cuban Anolis species. We then tested the explanatory value of the three factors outlined above for the observed genetic divergence among populations. Finally, to ensure that the effects of these factors were examined across a morphologically and ecologically diverse range of species, we analyzed a large number of species from Cuba.
Methods
Taxon sampling
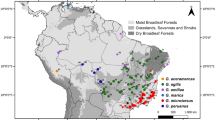
Field work was conducted during the wet seasons of 2008–2012 and was performed at 72 locations on the main island of the Cuban archipelago and on some smaller surrounding islands, such as Isla de la Juventud (Fig. 1). Sampling locations were selected to approximate known geographical ranges of each species based on previously published geographical distribution maps [32, 33]. Some individuals were preserved as voucher specimens and were deposited into the herpetological collection of the Graduate School of Life Sciences, Tohoku University, Japan. Tissue samples from other individuals were obtained by tail clipping and were stored in vials containing 90% ethanol. Locality data and GenBank accession numbers of samples from both this and a previous study [34] are summarized in Additional file 1: Table S1a. Ten Cuban species, Anolis agueroi, A. birama, A. delafuentei, A. garridoi, A. incredulus, A. juangundlachi, A. pigmaequestris, A. terueli, A. toldo, and A. vescus, were excluded from the present analyses. DNA sequences of four of these species (A. delafuentei, A. incredulus, A. oporinus, and A. toldo) were unavailable. The remaining species were not included in this study. Further, the inclusion of these species into the phylogeny did not alter the results. However, five species that had not been previously sequenced were added to the phylogeny (A. anfiloquioi, A. fugitivus, A. spectrum, A. litoralis, and A. ruibali). Numbers of samples and localities for each species are listed in Additional file 1: Table S1b.

Map of Cuba showing sample locations; Additional file 1: Table S1 contains more detailed information regarding these locations
DNA extraction
Total DNA was extracted from tail or leg muscle tissues from each individual using standard phenol-chloroform methods. The mitochondrial gene encoding NADH dehydrogenase subunit 2 (ND2; 1036 bp) and the two nuclear genes encoding zinc finger protein 521 (ZNF521; 634 bp) and fibrosin-like 1 (FBRSL1; 488 bp) were amplified and sequenced from all individuals using the primer sequences and amplification conditions described by A Cádiz, N Nagata, M Katabuchi, LM Diaz, LM Echenique-Diaz, HD Akashi, T Makino and M Kawata [34].
Phylogenetic analysis for intraspecific genetic distances
Two separate phylogenies were estimated using two different datasets. The first phylogeny was constructed for estimating intraspecific genetic distances as a proxy for genetic divergence among populations. The second was constructed for estimating divergence times and evaluating effects of species age on intraspecific genetic distances. The method for constructing the first phylogeny has been described in this section; the method for the second phylogeny has been described in the next section.
The first phylogeny was constructed using a dataset comprising 303 individuals belonging to 33 Cuban Anolis species. DNA sequences for species belonging to the trunk-ground ecomorph were obtained from a previous publication [34], and the phylogeny was constructed after selecting the localities on the main island of Cuba and excluding samples from the small surrounding islands. Two samples of Cuban Leiocephalus lizards (L. onaneyi and L. raviceps) were used as outgroups.
Genetic distances were estimated using a maximum likelihood (ML) tree that was generated from the combined data for the genes ND2, ZNF521, and FBRSL1. Tree topology was confirmed using Bayesian methods and prior to ML and Bayesian analyses, an appropriate model of sequence evolution and its parameters was determined using Kakusan 4.0 [35] The resulting GTR + Gamma+Invariant model [36] was applied to each of the regions based on the Akaike information criterion (AIC). ML analyses were performed using Treefinder [37] and robustness was validated using bootstrap analyses with 1000 replications. Bayesian analyses were performed to confirm ML topology using MrBayes version 3.1.2 [38]. Ten million generations of Markov Chain Monte Carlo (MCMC) were then performed with a sampling frequency of 1000. Subsequently, MCMC convergence was verified using Tracer 1.5 [39], and after discarding the first 2000 trees as burn-in, the remaining samples were used to estimate tree topology. Within-species pairwise genetic distances were calculated using the script distontree; http://www.fifthdimension.jp/products/distontree/. Generally, one or two (or in a small number of cases, three) samples per locality were used in the ML tree. However, when more than one sample from the same locality was analyzed, we calculated average genetic distances from all pairwise comparisons of samples from that location and from each of the other locations.
Phylogenetic analysis for divergence times
The second phylogeny for estimating divergence times was constructed by using the mitochondrial ND2 gene of Cuban and non-Cuban (from GenBank) Anolis species. Only mitochondrial ND2 gene was used because the nuclear genes were not available for non-Cuban Anolis species. The dataset for the phylogeny comprised samples from single individuals of all species from each location. These were selected from all of the collected Cuban Anolis species (51 species, 221 samples in total), and included 15 species (A. alfaroi, A. altitudinalis, A. anfiloquioi, A. baracoae, A. centralis, A. clivicola, A. guafe, A. guamuhaya, A. imias, A. litoralis, A. rejectus, A. ruibali, A. spectrum, A. vanidicus, and A. vermiculatus) that were collected in one location, but were not included in the ML tree. In addition, 47 non-Cuban Anolis species that were representative of all Caribbean islands (Jamaica, Hispaniola, Puerto Rico, Bahamas, Caymans, and Lesser Antilles) and of the continent (Central and South America), were selected using the most complete phylogeny available [40] as a reference, and included 2–3 species from each of their main clades. Finally, gene sequences for three Cuban species were downloaded from GenBank (A. macilentus, A. oporinus, and A. porcus) and were included in this phylogeny. We also used GenBank sequences for Basiliscus plumifrons and Polychrus acutirostris as outgroup species. Accession numbers for non-Cuban species are presented in Additional file 1: Table S2.
Divergence times were estimated using the relaxed clock model in BEAST 1.6.2. In these analyses, the ML tree generated by Treefinder was used as an initial tree for BEAST, and one run with 10 million generations of MCMC was performed. After MCMC, convergence was confirmed using Tracer v1.5, and the first 2000 trees were discarded as burn-in. We set one calibration point at the root of all species as 95 Mya according to a study reported by Nicholson et al. [41]. Fossil evidence is largely lacking for anoles, making the calibration of trees difficult. However, the following analyses do not require accurate ages of divergence because we compared the relative age of divergence among species.
Ecomorph classification
The ecomorph classification used here largely follows that of previous studies (e.g., [42, 43]), although we refined classifications for a group of 15 Cuban species that are currently considered unique anoles and therefore do not belong to any ecomorph class. We classified these Cuban unique anoles into unique-types 1, 2, 3, and 4 according to morphological, behavioral, and ecological characteristics, which may reflect dispersal abilities. Finally, we summarized the characteristics of each of these species groups (Additional file 1: Table S3), according to the studies by JB Losos [19] and L Rodríguez-Schettino [32].
Species used for the analysis
The present analyses were performed with independent monophyletic species. However, because the geographical ranges of A. ahli, A. barbatus, A. bartschi, A. confusus, A. cupeyalensis, A. fugitivus, A. quadriocelifer, and A. noblei are highly restricted, we sampled fewer than three closely-located areas and excluded data for these species from analyses.
For some species, populations from different regions formed different paraphyletic groups in both ML and Bayesian trees. Among these, A. allogus populations in the eastern region belonged to a separate clade from those in the western region, which belonged to the same clade as A. ahli ([34]; the present study). Thus, the separate paraphyletic clades of A. allogus were considered different species. Moreover, the average genetic divergence between localities tended to become larger when genetic distances between populations of these two clades were included. Hence, A. allogus populations were divided into eastern and west-central clades. Similarly, clades of A. allisoni and A. porcatus exhibited interspecific hybridization ([20]; the present study), and were divided into eastern and central, and eastern and west-central clades, respectively, and groups of these populations were considered separate species in analyses of the factors affecting intraspecific genetic divergences.
Populations of A. jubar were not monophyletic [34], and were therefore removed from analyses. In addition, the A. sagrei, sample 07_8_sag_W3, was not included in analyses because it was nested within the clade of A. bremeri, suggesting hybridization at locality W3. A similar decision was made for sample 41_7_bre_W8 of A. bremeri, which appeared to hybridize with A. sagrei at locality W8.
A total of 26 species was included in analyses of the factors that affect intraspecific genetic divergence. These included three species of the crown-giant ecomorph (A. equestris, A luteogularis, and A. smallwoodi), three species of the grass-bush ecomorph (A. alutaceus, A. cyanopleurus, and A. inexpectatus), five species of trunk-crown anoles (A. isolepis, Eastern A. allisoni, Central A. allisoni, Eastern A. porcatus, and West-Central A. porcatus), seven species of the trunk-ground ecomorph (Eastern A. allogus, West-Central A. allogus, A. bremeri, A. homolechis, A. mestrei, A. rubribarbus, and A. sagrei), three species of the twig ecomorph (A. alayoni, A. angusticeps, and A. guazuma), three species of unique-type 1 (A. argillaceus, A. pumilus, and A. loysianus), and two species of unique-type 2 (A. argenteolus and A. lucius).
Environments within species ranges
For each locality we extracted 19 bioclimatic variables from the WorldClim database (http://www.worldclim.org), as shown in Additional file 1: Table S4, and one vegetation variable (percent tree cover) based on satellite images of the entire globe from the MODIS sensor of Terra from the International Steering Committee for Global Mapping (https://globalmaps.github.io/ptc.html; Additional file 1: Table S4). Data of tree cover was used because the forest coverage could affect thermal environments that are important for distributions of anole species [34]. Data from WorldClim includes percent tree cover with 1-km resolution. Because anole samples were collected within a few kilometers of each sampling location, we averaged percent tree cover data with a 5-km resolution using ArcGIS ver. 10.0 so that the entire range of habitats was included for sampled populations. Values were then extracted for each variable at each location, and 20 environmental variables were normalized using principal components analysis (PCA). Subsequently, environmental dissimilarities (Euclidean distance) were calculated according to the variables on PCA axes 1–5 using R software (Additional file 1: Table S4).
Environmental heterogeneity between locations within species ranges was calculated and average environmental dissimilarity values were then calculated between species locations. These values were lower when the environments within the species ranges were homogeneous, and were increased with environmental differences between locations.
Geographic distances
Pairwise geographic distances were estimated using Geographic Distance Matrix Generator v1.2.3 [44] with samples from localities on the main island of Cuba. For each species, average values of all pairwise genetic and geographic distances (km) between locations were regarded as average genetic distances and average geographic distances for species, respectively. Because different species have different ranges and distributions, the number of sampling locations and the geographic distances between the locations varied. However, sampling locations were selected for the best feasible coverage of known geographic ranges of each species, leading to greater numbers of sampling locations for species with larger ranges. In some cases, sampling locations were selected to optimize detectability of species, and may have been affected by population density. Thus, sampling locations were generally representative of each species and average genetic distances for each species therefore represented characteristic geographic distances among major localities of species.
Because sampling locations were selected to cover the largest possible proportion of the known geographic ranges for each species, average geographic distances were highly correlated with range sizes (r = 0.8509, t = 7.9375, P = 3.625 × 10− 8), which were determined using a previously published distribution map [32]. Therefore, we omitted sizes of species ranges from our analysis to avoid multicollinearity in generalized least squares analyses. Although average geographic distances for each species were considered characteristic of geographic distances and were used as a proxy for species range, these sometimes depended on sampling locations. Thus, although average geographic distances were not considered explanatory, they were included as a covariate that potentially affects genetic distances. Thus, we determined predictors of average genetic divergence with consideration of average geographic distances.
Phylogenetic generalized least squares analyses
In phylogenetic generalized least squares (PGLS) analyses, we examine the effects of these factors on interspecific differences in genetic divergence within species, with consideration of the effects of phylogenetic constraints. In these analyses, average genetic distances among populations were entered as the response variable (estimated in section “Phylogenetic analysis for intraspecific genetic distances”), and average geographic distances between localities, ages of origin, average environmental dissimilarities and ecomorph types were included as explanatory variables. Average geographic distances between locations were included as a potential covariate of genetic distances. PGLS can consider the effect of phylogenetic constraints for the analysis and should include genetic distance matrix among species. For this purpose, the genetic distances among species were recorded and a phylogenetic tree was calculated from all individuals of the same species using the Maximum Composite Likelihood model in MEGA7 [45]. Because the genetic distance between species rather than individuals were used for removing phylogenetic constraints, we calculated genetic distances between species again using MEGA7. The resulting differences in composition biases among sequences were considered in previous evolutionary comparisons [46], and all positions containing gaps and missing data were eliminated. Subsequent PGLS analyses was conducted using the gls function in the “nmle” package of R (version 3.1.1), assuming a Brownian motion model of evolution.
Initially, PGLS analyses were performed using a full model that included all explanatory variables and potential interaction terms. Non-significant variables were then removed from the full model using a backward stepwise procedure with log-likelihood ratio tests, and the final model was constructed to include only significant variables and their interaction terms. Because PGLS could not be conducted using a model that only included the interaction terms, single variables that formed interaction terms were only included in the final model when they were significant in the full model. This final model was used to identify factors that are significantly associated with average genetic divergences within species.
Results
Phylogeny of Anolis species in Cuba
The present ML tree (Fig. 2) includes 303 individuals from 33 Cuban Anolis species and is more comprehensive than previous phylogenetic analyses of Cuban taxa. We incorporated a greater level of geographic sampling and a higher genetic resolution by including mitochondrial and nuclear markers. The topology of the ML tree was highly consistent with that generated using Bayesian inference (Additional file 2: Figure S1). Moreover, each of the present ecomorph classes formed monophyletic groups and similar results were obtained for groups of unique anoles, with the exception of the unique-type 2 species, which was polyphyletic. Accordingly, all twig anoles included in this tree (A. angusticeps, A. guazuma, and A. alayoni) were recovered as monophyletic species. These results were supported by the phylogenetic tree based only on the nuclear DNA tree (Additional file 2: Figure S2). Although previous studies classified these species as paraphyletic [40, 41], the present nodes were not well supported, warranting cautious conclusions of monophyly.

Maximum likelihood tree (50% majority consensus) based on three genes (2170 bp); 303 individuals from 33 Cuban Anolis species were included as the ingroup and two species from the genus Leiocephalus were included as the outgroup. Node supports are bootstrap percentages from maximum likelihood analyses; Bayesian posterior probabilities are shown only for the major clades. The figure has been split into parts A, B, and C, and some clades have been compressed to fit the full tree; CG, crown-giant; GB, grass-bush; T, twig; TC, trunk-crown; TG, trunk-ground; U1, unique-type 1; U2, unique-type 2; U4, unique-type 4; outgroup not shown
Phylogeny of Cuban and non-Cuban Anolis species and estimates of relative divergence times
We generated a phylogenetic tree with estimated relative divergence times from BEAST (Additional file 2: Figure S3), using 101 in-group species (54 Cuban and 47 non-Cuban species) and two out-group species. In agreement with the above ML analyses, all Cuban ecomorphs were recovered as monophyletic species with the exception of one twig species (A. guazuma), which appeared to be polyphyletic and was closely related to the clade comprising unique-type 1 and trunk-crown anoles.
Effects of average geographic distances, environmental heterogeneity, and species ages on interspecific differences in genetic divergence among populations
PGLS analyses showed that species age and geographic distance are significant predictors of average genetic distances between localities based on genetic distances that were calculated using nuclear and mitochondrial DNA (nDNA + mtDNA; Table 1a), or mtDNA only (Table 1b). Thus, older species tend to have larger genetic divergences (Fig. 3a) even when the significant effects of geographic distances are controlled. PGLS analyses with the full model also showed that environmental heterogeneity within species ranges and ecomorph types did not affect genetic distances among populations.

Relationship between average genetic divergences of species and species ages (a) and average geographic distances (b); residual regression values for species age and geographic variances vs. average genetic divergences were used
Discussion
Phylogeny of Cuban anoles
The phylogenies presented in this study follow the most comprehensive sampling of Cuban Anolis species to date. In particular, the species A. anfiloquioi, A. fugitivus, A. spectrum, A. litoralis, and A. ruibali had not been sequenced previously and were added to the phylogeny. Furthermore, lizard samples of most species were collected from their entire known geographic ranges, and we improved the molecular resolution using multiple genetic markers. Previous phylogenetic analyses of most Cuban species [40, 41, 47] were performed using mtDNA only, although some studies on smaller groups of Cuban species have included nuclear markers [20, 34, 48]. In particular, the most recent and comprehensive phylogenetic analysis of Anoles [49] includes more Cuban species than were included in the present study, but the DNA sampling accomplished here is much more extensive than in the previous study. They included 61 species from Cuba, but they used only morphological data for some species and lacked molecular data for 13 of them: A. toldo, A. litoralis, A. ruibali, A. terueli, A. juangundlachi, A. fujitivus, A. anfiloquioi, A. spectrum, A. vescus, A. pigmaequestris, A. delafuentei, A. incredulus, and A. birama. Thus, we accomplished a better DNA sampling of Anolis of Cuba than Poe et al. [49] because we sequenced five species (A. anfiloquioi, A. fujitivus, A. spectrum, A. litoralis, and A. ruibali) for the first time and we sampled most species from several localities.
Although the present phylogenetic relationships are mostly consistent with those of previous studies [49, 50], Cuban twig anoles were identified as a monophyletic group in our analysis (Fig. 2), indicating a more parsimonious evolution of Anolis ecomorphs on the island of Cuba, albeit with poor node support for this monophyly. Losos [19] similarly indicated that the separation of Cuban twig anoles into two clades is not strongly supported by mtDNA phylogeny. The monophyly of twig Anoles was observed only when using nuclear markers (Additional file 2: Figure S2) and combining both mitochondrial and nuclear DNA (Fig. 2) but not when using only mitochondrial DNA (Additional file 2: Figure S2). This explains why Cuban twig Anoles were recovered as a polyphyletic group in the phylogeny of Cuban and non-Cuban species (Additional file 2: Figure S2) and highlights the importance of using multiple molecular markers to uncover the evolutionary relationship of the Anolis lizards. Accordingly, the present data suggest that each well-defined ecomorph class (crown-giant, trunk-crown, trunk-ground, and twig) are monophyletic, with the exception of grass-bush anoles. In contrasting results, a group of Cuban anoles that were previously regarded as unique [19] was divided into four different groups according to ecological and morphological similarities, and, with the exception of unique-type 2 anoles (three rock-dwelling species), these groups were all monophyletic, as shown previously [40, 46].
The present tree of divergence time estimates (Additional file 2: Figure S3) were based on a previous study of anole divergence [40], in which origins of ancestral species were estimated for all anoles at 95 Mya. However, taxon compositions and geographic sizes of sampling areas of the present Cuban Anolis species differ from those in the study by Nicholson [40], and require consideration in comparisons of trees. In contrast, age estimations of the main nodes in our tree were generally lower than those reported previously [40], and estimates of dating may be inaccurate [51]; however, the absolute species age was not necessary for the present analysis as their effects on genetic diversity can be used for examining the effect of species ages. The extinction of sister species is responsible for overestimating the age of existing species. Here, we covered most of the existing species of Cuban anoles; therefore, we consider that extinction of sister species has had little effect, although it is possible that some species’ ages might be overestimated owing to the extinction or missing sampling of sister species.
Factors affecting interspecific differences in average genetic divergence among Cuban Anolis lizards
Our PGLS analyses suggest that species ages influence interspecific differences in genetic divergence among populations (Fig. 3), even after controlling for the effects of average geographic distances. In contrast, environmental heterogeneity within species ranges and ecomorph differences were not significantly associated with genetic divergence. Hence, older species have larger average genetic divergences, likely reflecting longer times to accumulate genetic divergence among populations. Accordingly, if gene flows between populations are very low, genetic divergence among populations will increase with time. In agreement, the present molecular phylogenic analysis indicates that, with few exceptions, individuals from the same or close locations form monophyletic groups, suggesting infrequent migration. Thus, when taxonomically older anole lizard species include older populations after divergence, species age becomes an important determinant of interspecific differences in genetic divergence among populations. Previous studies have related species age with geographical range sizes [11, 13, 14, 52], although several studies have reported unclear relationships between these variables [15, 53]. We identified a weak correlation between species age and range size (average geographic distances vs. species age, r = 0.0428, df = 24, P = 0.8356; PGLS, range = 61.43 age + 2354.09, t = 0.210, P = 0.8356), indicating that species range is not a covariate of species age.
A more accurate estimate of the genetic distance between populations requires intragenic variations using sufficient number of individuals within populations. To examine genetic divergence within species in our study, we examined 177 populations of 26 species; we usually used only one or few individuals for estimating genetic distances among populations. Both from a conservation viewpoint and because of technical difficulties, we could not collect sufficient numbers of samples within a population for all of the populations studied. Thus, our sampling strategy was to collect from several sampling locations to cover the largest possible proportion of the known geographic ranges for each species, and we collected randomly chosen one or few individuals from one population. Our aim was not to estimate the accurate values of genetic distance between populations, but to compare the relative degree of genetic divergence between populations among species. Our sampling procedure could reflect the average genetic divergence within species for species comparison. Further, in our studies, the examined genetic variations within populations would be not large. We used more than one sample (usually two) for 68 of 177 populations. The phylogeny of these individuals (Fig. 2) revealed that for 52 populations, the samples from the same populations were monophyletically related, so that genetic distances between samples within a population were smaller than those between the populations. Furthermore, seven of 16 populations wherein the samples within a population were paraphyletic; the samples from the same populations were genetically similar to those from geographically closer populations (~ < 50 km); therefore, our estimates of genetic distances were not largely affected by large intragenic population variation. Our study was based on the analysis of only few markers. Genome-wide data are essential for precise estimation of species age and genetic variations; further, we should also examine genetic divergence using genome-wide genetic markers in future studies.
We included geographic distance as a potential covariate of genetic distance in the present PGLS analyses, which indicated that geographic distance affects species genetic divergence. In addition, average geographic distances were highly correlated with species range sizes (r = 0.8509, t = 7.9375, P = 3.625 × 10− 8). Accordingly, PGLS analysis using species ranges instead of geographic distances showed that species ages and ranges significantly affect genetic divergence (Additional file 1: Table S5). Hence, although the effects of species range could not be separated from the effects of average measured geographic distances, species with larger species range may have larger genetic divergence. Fujisawa, Vogler and Barraclough [15] also showed that water beetle species with higher occupancy (i.e., a larger range size) were more genetically variable than species with lower occupancy, and suggested that species occupancy and range size are tightly correlated with population size, and that large population sizes are causal of genetic diversity [54, 55]. However, the relative contributions of population size and isolation-by-distance effects on genetic divergence were unclear in the present analyses.
Analyses of 19 climatic variables and percent tree cover found that species that inhabit more heterogeneous environments do not necessarily show higher levels of genetic divergence among populations based on 19 climatic variables and percent tree cover. In the present study, the number of individuals examined for some species was insufficient to conclude that environmental heterogeneity within species did not affect their genetic divergence; however, in some species, genetic divergence was lower, although environmental heterogeneity was high (e.g., Crown giant species such as A. luteogularis and A. smallwoodi). Thus, the results suggest that some Anolis species may not have adapted to local climatic differences, or local climate adaptations may not be related to divergence among populations when measured using neutral genetic markers. It has previously been hypothesized that Anolis lizard populations have adapted to environmental differences [19], wherein there should be variation in ecologically relevant traits among populations inhabiting different environments. For example, Anolis species that inhabit mountainous Lesser Antilles islands exhibit pronounced differences between populations, presumably following adaptations to different climates [56]. Moreover, in previous studies, geographic variations in ecologically relevant traits such as limb length, body shape, scalation, color, and body size have been associated with environmental variables such as temperature, humidity, and vegetation type [28, 57, 58]. However, Munoz et al. [59] demonstrated that color variations in A. marmoratus evolved in the absence of geographic isolation, and little or no genetic divergence was found among these populations using microsatellite loci. This could not eliminate the possibility that among Cuban Anolis species, local populations of a species can adapt to different climatic conditions without preventing substantial gene flow among the populations. Another study [6] showed that adaptive differences and selective pressures against dispersers play secondary roles in limiting gene flows between divergent habitats, constituting a small isolation-by-environment effect. However, these authors did not determine whether higher levels of environmental heterogeneity promote genetic divergence among habitats. The present results show that species that inhabit more heterogeneous environments do not necessarily have greater genetic divergence among populations.
In a study of inland snails, arboreal species had more divergent populations than ground-dwelling species [60], suggesting that species with different ecological niches have different patterns of genetic divergence among populations. Similarly, Anolis lizard species that belong to different ecomorphs may exhibit differing dispersal capacities due to divergence in morphology and microhabitat use, and these differences may be correlated with average genetic divergence. The present results could not find significant effects of ecomorph types on genetic divergence. However, we observed a tendency of crown-giant species to have lower genetic divergence than non-crown-giant species (0.0090 ± 0.0069 [SD] vs. 0.0467 ± 0.0207, respectively), suggesting that some ecomorphs’ characteristics affect genetic divergence. Accordingly, crown-giant species are clearly larger than other ecomorph species (mean male snout-vent length; 145.7 mm for crown-giant species and 34.8–59.18 for other ecomorph species), and may therefore cover greater distances and have greater dispersal ability. However, no significant effects of body size on genetic divergence were identified in PGLS analyses (t = − 1.1270, P = 0.2969). Further studies are required to consider whether other ecological or morphological characteristics affect genetic divergence among populations.
Phylogenies of trunk-ground and trunk-crown species usually show deep population divergence. For example, the trunk-ground species A. allogus is divided into two main clades, as shown herein and in a previous study [34]. Because one of these includes all eastern populations and the other comprises western and central populations, it has been suggested that these two clades should be considered as different species [34, 48]. Similarly, different paraphyletic clades have been identified in phylogenetic analyses of the two trunk-crown species A. allisoni and A. porcatus, again suggesting that eastern and western populations of A. porcatus should be distinguished at the species level based on genetic divergence [20]. Although we analyzed A. allisoni and A. porcatus as separate species, high genetic divergence was observed among populations of these clades, and deep population divergence was also observed among populations of A. homolechis, revealing unexpected levels of genetic variation within some Anolis species. Several previous studies have also showed deep interpopulation divergence among anoles from the Greater Antilles [20,21,22,23,24,25,26, 48] and Lesser Antilles [27,28,29,30,31], and in three Amazonian species [61]. Thus, our analyses indicate that the deep genetic divergences observed in several Anolis species could partly be driven by a longer isolation between populations.
Conclusion
In this study, we performed phylogenetic analyses of Cuban Anolis lizards using unprecedented numbers of species, geographic samples, and molecular markers. The present analyses showed more parsimonious evolution of Anolis ecomorphs within Cuba and identified twig anoles as a monophyletic group. Subsequent PGLS analyses confirmed that species age is positively correlated with species’ average genetic divergence among populations. Although previous studies have focused on factors affecting genetic divergence within species, the present study for the first time showed that species differences in genetic divergence could be largely affected by species age. The generalizations of the present study warrant further examination in studies of Caribbean and mainland anoles. Moreover, studies of genetic divergence among species that belong to similar ecomorph classes are required to confirm the present assertions on other Greater Antilles islands.
References
Coyne JA, Orr HA. Speciation. Sunderland: Sinauer Associates; 2004.
Bridle JR, Polechova J, Kawata M, Butlin RK. Why is adaptation prevented at ecological margins? New insights from individual-based simulations. Ecol Lett. 2010;13:485-94.
Wright S. Isolation by distance. Genetics. 1943;28:114–38.
Wang IJ, Summers K. Genetic structure is correlated with phenotypic divergence rather than geographic isolation in the highly polymorphic strawberry poison-dart frog. Mol Ecol. 2010;19(3):447–58.
Lee CR, Mitchell-Olds T. Quantifying effects of environmental and geographical factors on patterns of genetic differentiation. Mol Ecol. 2011;20(22):4631–42.
Wang IJ, Glor RE, Losos JB. Quantifying the roles of ecology and geography in spatial genetic divergence. Ecol Lett. 2013;16(2):175–82.
Brown JH, Maurer BA. Macroecology - the division of food and space among species on continents. Science. 1989;243(4895):1145–50.
Greenwood PJ. Mating systems, Philopatry and dispersal in birds and mammals. Anim Behav. 1980;28(Nov):1140–62.
Abellan P, Ribera I. Geographic location and phylogeny are the main determinants of the size of the geographical range in aquatic beetles. BMC Evol Biol. 2011;11:344.
Krug AZ, Jablonski D, Valentine JW. Species-genus ratios reflect a global history of diversification and range expansion in marine bivalves. Proc Biol Sci. 2008;275(1639):1117–23.
Paul JR, Morton C, Taylor CM, Tonsor SJ. Evolutionary time for dispersal limits the extent but not the occupancy of Species' potential ranges in the tropical plant genus Psychotria (Rubiaceae). Am Nat. 2009;173(2):188–99.
Swaegers J, Janssens SB, Ferreira S, Watts PC, Mergeay J, McPeek MA, Stoks R. Ecological and evolutionary drivers of range size in Coenagrion damselflies. J Evol Biol. 2014;27(11):2386–95.
Webb TJ, Gaston KJ. Geographic range size and evolutionary age in birds. Proc Biol Sci. 2000;267(1455):1843–50.
Wollenberg KC, Vieites DR, Glaw F, Vences M. Speciation in little: the role of range and body size in the diversification of Malagasy mantellid frogs. BMC Evol Biol. 2011;11:217.
Fujisawa T, Vogler AP, Barraclough TG: Ecology has contrasting effects on genetic variation within species versus rates of molecular evolution across species in water beetles. Proc Biol Sci. 2015; 282(1799).
Hedges SB. Caribherp: West Indian amphibians and reptiles (www.caribherp.org). Philadelphia: Temple University; 2018.
Williams EE. The origin of faunas. Evolution of lizard congeners in a complex island fauna: a trial analysis. Evol Biol. 1972;6:47–89.
Henderson RW, Powell R. Natural history of west Indian reptiles and amphibians. Gainesville: University Press of Florida; 2009.
Losos JB. Lizards in an evolutionary tree: ecology and adaptive radiation of anoles. California: University of California Press; 2009.
Glor RE, Gifford ME, Larson A, Losos JB, Schettino LR, Lara ARC, Jackman TR. Partial island submergence and speciation in an adaptive radiation: a multilocus analysis of the Cuban green anoles. Proc Biol Sci. 2004;271(1554):2257–65.
Glor RE, Kolbe JJ, Powell R, Larson A, Losos JB. Phylogenetic analysis of ecological and morphological diversification in Hispaniolan trunk-ground anoles (anolis cybotes group). Evolution. 2003;57(10):2383–97.
Glor RE, Losos JB, Larson A. Out of Cuba: overwater dispersal and speciation among lizards in the Anolis carolinensis subgroup. Mol Ecol. 2005;14(8):2419–32.
Jackman TR, Irschick DJ, De Queiroz K, Losos JB, Larson A. Molecular phylogenetic perspective on evolution of lizards of the Anolis grahami series. J Exp Zool. 2002;294(1):1–16.
Kolbe JJ, Glor RE, Schettino LR, Lara AC, Larson A, Losos JB. Multiple sources, admixture, and genetic variation in introduced Anolis lizard populations. Conserv Biol. 2007;21(6):1612–25.
Kolbe JJ, Glor RE, Schettino LRG, Lara AC, Larson A, Losos JB. Genetic variation increases during biological invasion by a Cuban lizard. Nature. 2004;431(7005):177–81.
Rodriguez-Robles JA, Jezkova T, Garcia MA. Evolutionary relationships and historical biogeography of Anolis desechensis and Anolis monensis, two lizards endemic to small islands in the eastern Caribbean Sea. J Biogeogr. 2007;34(9):1546–58.
Malhotra A, Thorpe RS. Parallels between island lizards suggests selection on mitochondrial-DNA and morphology. Proc Biol Sci. 1994;257(1348):37–42.
Malhotra A, Thorpe RS. The dynamics of natural selection and vicariance in the Dominican anole: patterns of within-island molecular and morphological divergence. Evolution. 2000;54(1):245–58.
Ogden R, Thorpe RS. Molecular evidence for ecological speciation in tropical habitats. Proc Natl Acad Sci USA. 2002;99(21):13612–5.
Schneider CJ. Distinguishing between primary and secondary intergradation among morphologically differentiated populations of Anolis marmoratus. Mol Ecol. 1996;5(2):239–49.
Thorpe RS, Stenson AG. Phylogeny, paraphyly and ecological adaptation of the colour and pattern in the Anolis roquet complex on Martinique. Mol Ecol. 2003;12(1):117–32.
Rodríguez-Schettino L. The iguanid lizards of Cuba. Gainesville: University of Florida Press; 1999.
Schwartz A, Henderson RW. Amphibians and reptiles of the West Indies: descriptions, distributions, and natural history. Gainesville: University of Florida Press; 1991.
Cádiz A, Nagata N, Katabuchi M, Diaz LM, Echenique-Diaz LM, Akashi HD, Makino T, Kawata M. Relative importance of habitat use, range expansion, and speciation in local species diversity of Anolis lizards in Cuba. Ecosphere. 2013;4:7.
Tanabe AS. Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour. 2011;11(5):914–21.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–26.
Jobb G, von Haeseler A, Strimmer K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004;4:18.
Ronquist F, Huelsenbeck JP. Mrbayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–4.
Rambaut A, Drummond AJ. Tracer v1.5. http://beast.bio.ed.ac.uk/Tracer; 2009.
Nicholson KE, Crother BI, Guyer C, Savage JM. It is time for a new classification of anoles (Squamata: Dactyloidae) (vol 3477, pg 1, 2012). Zootaxa. 2012(3480):88–8.
Nicholson KE, Glor RE, Kolbe JJ, Larson A, Hedges SB, Losos JB. Mainland colonization by island lizards. J Biogeogr. 2005;32(6):929–38.
Beuttell K, Losos JB. Ecological morphology of Caribbean anoles. Herpetol Monogr. 1999;13:1–28.
Johnson MA, Leal M, Schettino LR, Lara AC, Revell LJ, Losos JB. A phylogenetic perspective on foraging mode evolution and habitat use in west Indian Anolis lizards. Anim Behav. 2008;75:555–63.
Ersts PJ. Geographic distance matrix generator(version 1.2.3). American Museum of Natural History, Center for Biodiversity and Conservation. New York: American Museum of Natural History; 2015.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Tamura K, Kumar S. Evolutionary distance estimation under heterogeneous substitution pattern among lineages. Mol Biol Evol. 2002;19(10):1727–36.
Jackman TR, Larson A, de Queiroz K, Losos JB. Phylogenetic relationships and tempo of early diversification in Anolis lizards. Syst Biol. 1999;48(2):254–85.
Knouft JH, Losos JB, Glor RE, Kolbe JJ. Phylogenetic analysis of the evolution of the niche in lizards of the Anolis sagrei group. Ecology. 2006;87(7):S29–38.
Poe S, de Oca ANM, Torres-Carvajal O, de Queiroz K, Velasco JA, Truett B, Gray LN, Ryan MJ, Kohler G, Ayala-Varela F, et al. A phylogenetic, biogeographic, and taxonomic study of all extant species of Anolis (Squamata; Iguanidae). Syst Biol. 2017;66(5):663–97.
Paradis E. Analysis of Phylogenetics and evolution with R. second ed. New York: Springer; 2012.
Castaneda MD, Sherratt E, Losos JB. The Mexican amber anole, Anolis electrum, within a phylogenetic context: implications for the origins of Caribbean anoles. Zool J Linn Soc. 2014;172(1):133–44.
Jablonski D. Heritability at the species level - analysis of geographic ranges of cretaceous mollusks. Science. 1987;238(4825):360–3.
Mora C, Treml EA, Roberts J, Crosby K, Roy D, Tittensor DP. High connectivity among habitats precludes the relationship between dispersal and range size in tropical reef fishes. Ecography. 2012;35(1):89–96.
Blackburn TM, Cassey P, Gaston KJ. Variations on a theme: sources of heterogeneity in the form of the interspecific relationship between abundance and distribution. J Anim Ecol. 2006;75(6):1426–39.
Gaston KJ, Blackburn TM, Greenwood JJD, Gregory RD, Quinn RM, Lawton JH. Abundance-occupancy relationships. J Appl Ecol. 2000;37:39–59.
Thorpe RS, Barlow A, Malhotra A, Surget-Groba Y. Widespread parallel population adaptation to climate variation across a radiation: implications for adaptation to climate change. Mol Ecol. 2015;24(5):1019–30.
Calsbeek R, Knouft JH, Smith TB. Variation in scale numbers is consistent with ecologically based natural selection acting within and between lizard species. Evol Ecol. 2006;20(4):377–94.
Thorpe RS, Malhotra A, Stenson AG, Reardon JT. Adaptation and speciation in Lesser Antillean anoles. In: Dieckmann U, Doebeli M, JAJ M, Tautz D, editors. Adaptive Speciation. Cambridge: Cambridge University Press; 2004. p. 322–44.
Munoz MM, Crawford NG, Mcgreevy TJ, Messana NJ, Tarvin RD, Revell LJ, Zandvliet RM, Hopwood JM, Mock E, Schneider AL, et al. Divergence in coloration and ecological speciation in the Anolis marmoratus species complex. Mol Ecol. 2013;22(10):2668–82.
Davison A, Chiba S. Contrasting response to Pleistocene climate change by ground-living and arboreal Mandarina snails from the oceanic Hahajima archipelago. Philos Trans R Soc B. 2008;363(1508):3391–400.
Glor RE, Vitt LJ, Larson A. A molecular phylogenetic analysis of diversification in Amazonian Anolis lizards. Mol Ecol. 2001;10(11):2661–8.
Acknowledgments
We thank Jonathan B. Losos for his constructive comments on earlier versions of the manuscript and give special thanks to Chiemi Saito, who has offered continuous support to AC in recent years. We also thank Gaku Takimoto, Wataru Anzai, Lainet García, Ariatna Linares, Mey L. Reytor, and the Unidad de Medio Ambiente de Guantánamo, particularly Yamilka Joubert, Gerardo Begué, and Yonder Verdecia, for their assistance during fieldwork. Noe Matsushima provided laboratory assistance. Ansel Fong and the Museo Nacional de Historia Natural de Cuba kindly provided specimens for phylogenetic analysis.
Funding
MK was supported by a Grant-in-Aid for Scientific Research (22405008 and 25304016) from the Japan Society for the Promotion of Science (JSPS).
Availability of data and materials
DNA data have been deposited in GenBank. Locality data and GenBank accession numbers of samples are summarized in Additional file 1: Table S1.
Author information
Authors and Affiliations
Contributions
AC and MK conceived the ideas and designed the study; AC, LM, HA, and LE collected samples and data. AC, NN, and TM conducted phylogenetic analysis and AC and YS and KM conducted statistical analysis. AC and MK wrote the article. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Collection and exportation permits were provided by the Centro de Inspección y Control Ambiental (CICA) of the Agencia de Medio Ambiente de Cuba. No experiments or procedures involving animals were performed in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. (a) Location data and GenBank accession numbers of the Cuban specimens used in this study. (b) Regional sampling data. Table S2. GenBank sequence accession numbers of the non-Cuban specimens used in this study. Table S3. Description of Unique Type in Cuba Anoles. Table S4. Environmental variables for each locality. Table S5. Results of a phylogenetic generalized least squares (PGLS) analysis for the final model using both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) (a) and only mtDNA (b). (DOC 627 kb)
Additional file 2:
Figure S1. Bayesian tree (50% majority consensus) based on three genes using 303 individuals from 33 Cuban Anolis species and two species from the genus Leiocephalus. Figure S2. Contrasting Bayesian phylogenies for nuclear DNA (based on ZNF521 and FBRSL1) and mtDNA (based on ND2) for Cuban anoles. Figure S3. Phylogenetic tree with estimates of divergence times based on the mitochondrial gene ND2 (1036 bp). (DOC 8930 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cádiz, A., Nagata, N., Díaz, L.M. et al. Factors affecting interspecific differences in genetic divergence among populations of Anolis lizards in Cuba. Zoological Lett 4, 21 (2018). https://doi.org/10.1186/s40851-018-0107-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40851-018-0107-x