
Abstract
Background
The Emei Shan Liocichla (Liocichla omeiensis) is an endemic bird species to southwestern China with a small geographic range. However, little was known about the genetic status of this threatened species.
Methods
We applied restriction-site-associated DNA sequencing (RAD-Seq) for rapid mass identification of microsatellite markers of the Emei Shan Liocichla.
Results
A total of 11,564 microsatellite sequences were obtained, 600 random loci were designed for screening and 24 polymorphic microsatellite loci were selected for further validation. The average allele number, average observed heterozygosity and average expected heterozygosity were relatively low in our samples, which were 6.08, 0.6618 and 0.7048, respectively, indicating that the Emei Shan Liocichla might have lost some genetic diversity. Further analyses suggested that the populations distributed on two mountains (Daxiangling and Xiaoliangshan) showed a modest degree of genetic differentiation.
Conclusions
These novel microsatellite markers provided valuable preliminary knowledge regarding the genetic status of the Emei Shan Liocichla and can be useful in further studies, as well as in the management and conservation of this species.
Similar content being viewed by others


Background
Genetic diversity is crucial for the sustainability of species, especially for threatened species (Meffe and Carroll 1994). Habitat fragmentation, a common cause of endangering threatened species, can lead to reductions in the population size and gene flow among patches, which leads to a loss of genetic diversity and an increase in inbreeding (Frankham et al. 2002). The loss of genetic diversity associated with inbreeding has two important consequences: it reduces the ability of populations to adapt to environmental/climate change, and it can result in a direct and immediate loss in fitness through inbreeding depression (Keller and Waller 2002; Armstrong and Seddon 2008; Jamieson 2011). It is, therefore, necessary to assess the genetic diversity and inbreeding level, and to determine whether a population is genetically fragmented before undertaking the genetic management actions for threatened species, which are important in biodiversity conservation (Frankham et al. 2002).
Microsatellites have a higher mutation rate compared to mitochondrial DNA and nuclear introns (Ryman and Leimar 2008), a feature that can provide a powerful tool for detecting genetic differences, especially in small and fragmented populations (Li et al. 2002; Duncan et al. 2016). Microsatellite markers have been widely applied in molecular ecology and conservation genetic studies since the 1990s (Bruford and Wayne 1993; Jarne and Lagoda 1996; Chapuis et al. 2015; Faria et al. 2016). Although cross-species amplification works for a few closely related taxa (Dawson et al. 2010; Gu et al. 2012), the major drawback of microsatellites is that they need to be independently isolated from most species (Sudheer et al. 2010). Most microsatellites must be identified by screening genomic libraries with appropriate probes or enrichment protocols, which are inefficient and time-consuming (Zane et al. 2002). Whereas the Illumina next generation sequencing (NGS) platform, which can produce moderately long paired-end reads (up to 150 bp with the HiSeq 2500 platform), is an effective method for obtaining reliable microsatellites, even for birds that have few microsatellite loci (Castoe et al. 2012). The restriction-site-associated DNA (RAD) method (Miller et al. 2007) with Illumina sequencing (RAD-Seq) can further reduce the cost and improve the average depth per locus (Baird et al. 2008). Unlike other methods for generating genome-wide data, RAD-Seq does not require any prior genomic information of the taxa being studied (Andrews et al. 2016). To date, several studies have shown that paired-end RAD-Seq is a powerful tool for obtaining microsatellites in both model and non-model species (Miller et al. 2007; Pfender et al. 2011).
The vulnerable (VU) Emei Shan Liocichla (Liocichla omeiensis) is an endemic passerine bird found on Daxiangling Mountain and Xiaoliangshan Mountain in southwestern China. It lives on the edges or gaps of natural broadleaf forests between an elevation of 1450 and 2150 m (Fu et al. 2011). This bird is resident and moves vertically between seasons (Fu and Zhang 2011). Additionally, the Emei Shan Liocichla is socially monogamous and protects its territory continually throughout the year, with a territory range of approximately 10,000 m2 (Fu, unpublished data). However, the population of the Emei Shan Liocichla is suspected to be declining at a moderate rate, which is in line with the accelerated habitat loss and degradation within its range (BirdLife International 2017). The natural broadleaf forests in its habitat have been cleared or replaced by non-native coniferous forests and tea plantations, which might lead to habitat fragmentation as a previous study has shown the Emei Shan Liocichla did not use these types of vegetation (Fu et al. 2011). Habitat loss and fragmentation are regarded as the major factors contributing to the population decline in many bird species (Ribon et al. 2003; Gill 2007). In addition, during our sampling period, we witnessed a number of captures of these birds for sale, which will also damage the population. Although conservation actions have been taken since 1999, little is known about the genetic status of this vulnerable species and its populations. Therefore, to carry out genetic diagnoses before undertaking genetic management actions (Frankham et al. 2002), a set of suitable genetic markers, such as microsatellites, are urgently needed.
In this study, we utilized Illumina paired-end RAD-Seq to isolate highly polymorphic microsatellites of the Emei Shan Liocichla that can be useful for further studies, and tried to understand some preliminary genetic status to help the management and conservation of this vulnerable species using our incomplete sampling by answering the following questions: (1) how much genetic diversity is there in our sampling populations? (2) are the populations suffering from inbreeding? (3) are the populations from two mountains already genetically fragmented? and (4) what we can do for conservation of the Emei Shan Liocichla?
Methods
Sample collection and DNA purification
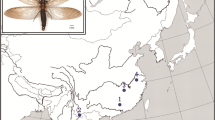
Fifty-one samples containing 38 blood and 13 tissue samples, were collected from six sites distributed in Daxiangling Mountain and Xiaoliangshan Mountain located in southwestern Sichuan, China (Fig. 1). Blood samples were collected from the brachial vein and stored in absolute ethanol. All the captured individuals were released immediately after the blood samples were obtained. Tissue samples were collected opportunistically from individuals that died naturally and dead chicks in nests. Permission for the sampling was granted by the Sichuan Forestry Department and the Laojunshan National Nature Reserve of Sichuan Province, China. The samples were frozen in the field and preserved at −80 °C in the lab. Total genomic DNA was extracted using TIANamp Genomic DNA Kit DP304-2 (Tiangen, China) following the manufacturer’s recommendations.

Sample locations of the Emei Shan Liocichla (Liocichla omeiensis)
RAD-Seq and microsatellite characterization
In total, 3 µg of genomic DNA from a single male was sent to Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) for the DNA library preparation and sequencing to obtain sequencing data for microsatellite screening. We used P1 and P2 adaptors for the restriction enzyme sites to construct a DNA sequencing library with a size range of 300–700 bp. Paired-end 125 bp reads were obtained from separate lanes of the Hiseq 2500 Genome Analyzer. Poor-quality reads were cleaned and adapter reads were trimmed using Trimmomatic 0.36 (Bolger et al. 2014) with default settings. Reads containing RAD tags were clustered by different RAD tags using cd-hit-est (Li and Durbin 2009), and RAD tags containing 10‒400 reads were considered repetitive and removed. The remaining paired-end reads from each RAD site were then sent to Velvet (Zerbino and Birney 2008) to assemble contigs with the following parameters settings: VelvetOptimiser.pl -s 23 -e 31 -x 4. An SR search was developed by Novogene Bioinformatics Technology (Beijing, China) to detect the microsatellites from contigs using the following criteria: 2–6-bp motifs, longer than 12 bp, and the distance between two microsatellites was greater than 12 bp.
Primer design and microsatellite evaluation
We identified 11,564 putative microsatellite sequences after excluding compound and interrupted microsatellites. We randomly designed 600 pairs of primers ranging in length from 12 to 36 bp using Primer Premier 5 (Lalitha 2000). The predicted PCR products included trimers, quadmers, pentamers and hexamers. PCR was performed using these primers on four separate Emei Shan Liocichla DNA samples. PCR was conducted in 10 μL reaction volumes containing ~20 ng of genomic DNA, 1 μL of 20 μM of each primer, and 3‒5 μL 2× Taq polymerase (Tiangen, China). Following a denaturation step of 5 min at 95 °C, the PCR mixture was subjected to 30 cycles of 94 °C for 30 s, Tm for 30 s, and 72 °C for 45 s, followed by 72 °C for 10 min and storage at 4 °C. A total of 99 primer pairs were chosen with clear straps, correct placement, and no non-specific amplification by agarose gel electrophoresis. Each locus was examined in eight Emei Shan Liocichla individuals by sequencing the PCR products (Tsingke, Beijing), and MEGA 7.0 (Kumar et al. 2016) was used to assess polymorphisms at each locus. In total, 47 microsatellite loci with one or two alleles were excluded, and each of the remaining 52 microsatellite loci was labelled with one fluorescent dye (FAM, HEX, or TAMRA) to the forward primers. We then amplified all these loci in 51 samples and performed genotyping using Genemapper 4.0 (Applied Biosystems).
Statistical and genetic data analysis
The number of alleles per locus (N a), observed heterozygosity (H o), expected heterozygosity (H e), and polymorphic information content (PIC) were computed via CERVUS 3.0 (Marshall et al. 1998). Tests for deviations from the Hardy–Weinberg equilibrium (HWE), linkage-disequilibrium (LD), and the inbreeding coefficient (F IS) were conducted by Arlequin 3.5.2.2 (Excoffier and Lischer 2010). The resulting p values were adjusted by applying a sequential Bonferroni correction using SGoF+ 3.8 (Carvajal-Rodriguez and de Uña-Alvarez 2011). A maximum-likelihood estimate of the frequency of null alleles and allelic dropout was calculated for each locus using Micro-Checker 2.2.3 (Van Oosterhout et al. 2004). Genetic differential index (F ST) for the populations between two mountains was estimated in GENETIX 4.05.2 using 1000 bootstraps to calculate significance (Belkhir et al. 2004). We used BayesAss 3.03 to estimate rates of recent immigration between populations from two mountains (Wilson and Rannala 2003). Model parameters were set to default values, and the MCMC was run for 4,000,000 iterations, with a burn-in of 1,000,000 iterations and a sampling frequency of 2000. The chromosome location of each microsatellite was obtained by comparison with the Zebra Finch (Taeniopygia guttata) genome (http://www.ncbi.nlm.nih.gov/genome/seq/BlastGen.cgi?taxid=59729).
Results
Identification of microsatellite loci
A total of 61,704,680 NGS reads (7.18 G bp) were generated in this study. After removing the adapter-related, low quality, and duplicated reads, 56,901,588 reads remained (92.22%). We obtained 24,408,786 clean reads after RAD tag clustering. These reads were assembled into 973,893 contigs. The average length and the N50 of contigs were approximately 361 and 510 bp, respectively. The average GC content of retained contigs was 40.41% and the average sequencing depth was 17.03.
In total, 11,564 putative microsatellites contained 3152 di-nucleotides, 6030 tri-nucleotides, 1886 tetra-nucleotides, 395 penta-nucleotides, and 101 hexa-nucleotides. Within the 600 random microsatellite-containing sequences, there were 119 tri-nucleotides, 250 tetra-nucleotides, 136 penta-nucleotides, and 95 hexa-nucleotides.
Based on the criteria that a microsatellite locus must contain more than two alleles, high heterozygote peak height ratios, minimal stuttering and split peaks (Guichoux et al. 2011), 24 microsatellite primers (Table 1) were selected to genotype 51 Emei Shan Liocichla samples.
Genetic characters of the Emei Shan Liocichla samples
The N a ranged from 4 to 10 with an average of 6.08. H o and H e varied from 0.3922 to 0.9020 (mean = 0.6617) and 0.4628 to 0.8464 (mean = 0.7047), respectively. PIC values ranged from 0.4280 to 0.8180 with an average of 0.6546. After the Bonferroni correction, five loci exhibited deviations from HWE (LO131, LO299, LO313, LO514 and LO55), and two pairwise loci exhibited linkage disequilibrium (LO131 and LO55, LO313 and LO55). Therefore, we removed these five potential problematic loci in following analyses. The overall F IS value of the remaining 19 loci was −0.00976, significantly different from zero (p = 0.0020). Estimated frequencies of null alleles per locus ranged from 0 to 0.1739, and there is no allelic dropout in each locus. The detailed characteristics of all individuals at the 24 loci are summarized in Table 1.
The overall F ST between populations from Daxiangling and Xiaoliangshan is 0.022 (p < 0.05). The recent migration rate calculated from BayesAss was 0.3128 from populations in Daxiangling to populations in Xiaoliangshan, and 0.0291 vice versa.
Discussion
Our results identified 24 novel microsatellites of the Emei Shan Liocichla using Illumina paired-end RAD-Seq strategy. The higher average depth per locus yielded by RAD-Seq ensured the accuracy of these microsatellites (Andrews et al. 2016).
PIC is the most widely applied formula to measure the information content of molecular markers (Nagy et al. 2012). According to the criteria of Botstein et al. (1980), 21 of these 24 microsatellite loci characterized by RAD sequencing showed a high level of polymorphism (PIC > 0.5). Other researchers like Huang et al. (2015) and Zhang et al. (2015) who have used other NGS methods to identify microsatellites within threatened bird species have observed similar polymorphism levels. Therefore, the use of RAD sequencing strategy to identify microsatellite is economically appealing and effective (Barchi et al. 2011).
The levels of microsatellite genetic diversity in the Emei Shan Liocichla shown in our results are lower than those in the non-threatened Liocichla steerii (McKay et al. 2010) (Table 2). Although the comparison between different microsatellite loci can be problematic, the N a in L. steerii (9–51) (McKay et al. 2010) are much higher than in Emei Shan Liocichla (4–10, Table 2), which could still imply that the Emei Shan Liocichla might have lost some genetic diversity. Additionally, the frequency of null alleles was low for most loci (r < 0.05), suggesting that the effective population size for the Emei Shan Liocichla might be low (Chapuis and Estoup 2007), consistent with the low number of mature individuals estimated (1500‒7000) (BirdLife International 2017).
After removing HWE-deviated and LD loci, the overall F IS showed negative values in all samples (Table 2), which means that the populations of the Emei Shan Liocichla do not exhibit inbreeding. It has been proposed that sexual dimorphism, which implies potential mating preference, can be a possible mechanism to avoid inbreeding (Tregenza and Wedell 2002). The males of Emei Shan Liocichla differ from females in having conspicuous orange-red on flight-feather bases and tips (Collar et al. 2016). In contrast, L. steerii show less sexual dimorphism between males and females, and their F IS valve was 0.026 (Table 2) even in such large populations. Therefore, the low-level inbreeding in the Emei Shan Liocichla may be due to strong sexual selection, an explanation that needs to be further investigated.
The global F ST between populations from Daxiangling and Xiaoliangshan was low (0.022), but highly significant, which indicate that there is a modest degree of genetic differentiation between populations from these two mountains. However, the recent migration rate analysis indicated there was relatively high contemporary gene flow from Daxiangling populations to Xiaoliangshan populations (0.3128), suggesting the populations are not strictly isolated yet. It is worth noting that the recent migration rate was highly asymmetrical, implying that a source-sink population structure exists in the Emei Shan Liocichla. Worse yet, the natural forests within its habitat have been cleared or replaced by tea plantations, and although China’s Grain-for-Green Program has been in effect since 1999, the restored forests between these two mountains are mostly monoculture forest and semi-mixed forest (Hua et al. 2016), which may be unsuitable for bird dispersal (Twedt et al. 2002). Therefore, although the populations exhibit low levels of genetic differentiation and moderate unidirectional gene flow now, still the long-term separation between these populations and unsuitable habitat between the two mountains may increase the risk of genetic fragmentation.
Conclusions
Based on the aforementioned results, 19 neutral microsatellites, with considerably high polymorphisms, were explored in the Emei Shan Liocichla by Illumina paired-end RAD-Seq. This set of de novo nuclear markers, combined with other molecular markers, such as mitochondrial DNA and nuclear introns, would facilitate further studies on population demographics, phylogeography and paternity analysis in vulnerable Emei Shan Liocichla and might be employed across species within the Liocichla genus, such as the critically endangered Bugun Liocichla (Liocichla bugunorum). In our genetic diagnoses, the Emei Shan Liocichla showed decreased genetic diversity, and a modest degree of genetic differentiation between populations, although they showed no inbreeding yet. These results indicate that if we conserve this species properly, before the emergence of strict loss of genetic diversity and inbreeding, we may still be able to prevent the population from becoming fully genetically fragmented.
References
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92.
Armstrong DP, Seddon PJ. Directions in reintroduction biology. Trends Ecol Evol. 2008;23:20–5.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3:e3376.
Barchi L, Lanteri S, Portis E, Acquadro A, Valè G, Toppino L, Rotino GL. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genom. 2011;12:1–9.
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. GENETIX 4.05, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier (France); 2004.
BirdLife International. Liocichla omeiensis. The IUCN Red List of Threatened Species 2017. Cambridge: BirdLife International; 2017.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Human Genet. 1980;32:314–31.
Bruford MW, Wayne RK. Microsatellites and their application to population genetic studies. Curr Opin Genet Dev. 1993;3:939–43.
Carvajal-Rodriguez A, de Uña-Alvarez J. Assessing significance in high-throughput experiments by sequential goodness of fit and q-value estimation. PLoS ONE. 2011;6:e24700.
Castoe TA, Poole AW, de Koning APJ, Jones KL, Tomback DF, Oyler-McCance SJ, Fike JA, Lance SL, Streicher JW, Smith EN, Pollock DD. Rapid microsatellite identification from illumina paired-end genomic sequencing in two birds and a snake. PLoS ONE. 2012;7:e30953.
Chapuis M-P, Estoup A. Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol. 2007;24:621–31.
Chapuis MP, Plantamp C, Streiff R, Blondin L, Piou C. Microsatellite evolutionary rate and pattern in Schistocerca gregaria inferred from direct observation of germline mutations. Mol Ecol. 2015;24:6107–19.
Collar N, Robson C, de Juana E. Grey-cheeked Liocichla (Liocichla omeiensis). In: del Hoyo J, Elliott A, Sargatal J, Christie DA, de Juana E, editors. Handbook of the birds of the world alive. Barcelona: Lynx Edicions; 2016.
Dawson DA, Horsburgh GJ, Küpper C, Stewart IRK, Ball AD, Durrant KL, Hansson B, Bacon IDA, Bird S, Klein Á, Krupa AP, Lee JW, Martín-Gálvez D, Simeoni M, Smith G, Spurgin LG, Burke T. New methods to identify conserved microsatellite loci and develop primer sets of high cross-species utility—as demonstrated for birds. Mol Ecol Resour. 2010;10:475–94.
Duncan CJ, Worth JRP, Jordan GJ, Jones RC, Vaillancourt RE. Genetic differentiation in spite of high gene flow in the dominant rainforest tree of southeastern Australia, Nothofagus cunninghamii. Heredity. 2016;116:99–106.
Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.
Faria J, Pita A, Rivas M, Martins GM, Hawkins SJ, Ribeiro P, Neto AI, Presa P. A multiplex microsatellite tool for conservation genetics of the endemic limpet Patella candei in the Macaronesian archipelagos. Aquat Conserv. 2016;26:775–81.
Frankham R, Briscoe DA, Ballou JD. Introduction to conservation genetics. Cambridge: Cambridge University Press; 2002.
Fu Y, Zhang Z. The influence of abnormal low-temperature of spring and summer on the breeding of Omei Shan Liocichla (Liocichla omeiensis). Beijing Norm Univ (Nat Sci). 2011;47:292–5 (in Chinese).
Fu Y, Dowell SD, Zhang Z. Breeding ecology of the Emei Shan Liocichla (Liocichla omeiensis). Wilson J Ornithol. 2011;123:748–54.
Gill FB. Ornithology. New York: W.H. Freeman & Company; 2007.
Gu LY, Liu Y, Wang N, Zhang ZW. A panel of polymorphic microsatellites in the Blue Eared Pheasant (Crossoptilon auritum) developed by cross-species amplification. Chin Birds. 2012;3:103–7.
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O, Lepoittevin C, Malausa T, Revardel E, Salin F, Petit RJ. Current trends in microsatellite genotyping. Mol Ecol Resour. 2011;11:591–611.
Hua F, Wang X, Zheng X, Fisher B, Wang L, Zhu J, Tang Y, Douglas WY, Wilcove DS. Opportunities for biodiversity gains under the world’s largest reforestation programme. Nat Commun. 2016;7:12717.
Huang J, Zhu D, Song X, Chen B, Zeng C, Moermond T, Zhang X, Yue B. High-throughput microsatellite markers discovery for the Sichuan Hill Partridge (Arborophila rufipectus) and assessment of genetic diversity in the Laojunshan population. Biochem Syst Ecol. 2015;60:266–72.
Jamieson IG. Founder effects, inbreeding, and loss of genetic diversity in four avian reintroduction programs. Conserv Biol. 2011;25:115–23.
Jarne P, Lagoda PJL. Microsatellites, from molecules to populations and back. Trends Ecol Evol. 1996;11:424–9.
Keller LF, Waller DM. Inbreeding effects in wild populations. Trends Ecol Evol. 2002;17:230–41.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Lalitha S. Primer premier 5. Biotech Softw Internet Rep. 2000;1:270–2.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li Y, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11:2453–65.
Marshall TC, Slate J, Kruuk LEB, Pemberton JM. Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol. 1998;7:639–55.
McKay BD, Mays HL, Peng Y-W, Kozak KH, Yao C-T, Yuan H-W. Recent range-wide demographic expansion in a Taiwan endemic montane bird, Steere’s Liocichla (Liocichla steerii). BMC Evol Biol. 2010;10:71.
Meffe GK, Carroll CR. Principles of conservation biology. Sunderland: Sinauer; 1994.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17:240–8.
Nagy S, Poczai P, Cernák I, Gorji AM, Hegedűs G, Taller J. PICcalc: an online program to calculate polymorphic information content for molecular genetic studies. Biochem Genet. 2012;50:670–2.
Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122:1467–80.
Ribon R, Simon JE, Theodoro De Mattos G. Bird extinctions in Atlantic forest fragments of the Viçosa region, southeastern Brazil. Conserv Biol. 2003;17:1827–39.
Ryman N, Leimar O. Effect of mutation on genetic differentiation among nonequilibrium populations. Evolution. 2008;62:2250–9.
Sudheer PDVN, Mastan SG, Rahman H, Prakash CR, Singh S, Reddy MP. Cross species amplification ability of novel microsatellites isolated from Jatropha curcas and genetic relationship with sister taxa. Mol Biol Rep. 2010;38:1383–8.
Tregenza T, Wedell N. Polyandrous females avoid costs of inbreeding. Nature. 2002;415:71–3.
Twedt DJ, Wilson RR, Henne-Kerr JL, Grosshuesch DA. Avian response to bottomland hardwood reforestation: the first 10 years. Restor Ecol. 2002;10:645–55.
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. 2004;4:535–8.
Wilson GA, Rannala B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics. 2003;163:1177–91.
Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review. Mol Ecol. 2002;11:1–16.
Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9.
Zhang L, Zhang ZH, Shen FJ, Hou R, Zhang WP, Liu YL, Tu KY, Yang AL. Identification and characterization of polymorphic microsatellite loci in the red-crowned crane. Genet Mol Res. 2015;14:15169–76.
Authors’ contributions
AY designed primers, conducted genetic analyses and led efforts to draft the manuscript. DC analyzed the data, interpreted the outcomes and helped draft the manuscript. PW and YF helped with acquired samples. ZZ and YF conceived the ideas, improved the manuscript and directed the research. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 31272330) to YF and the National Key Programme of Research and Development, Ministry of Science and Technology (2016YFC0503200) to ZZ. We would like to thank Mr. Jianzhi Zhang for his help in sample collection, and Mr. Yinong Liu for his help in figure preparation. We appreciate Dr. Rowden for his help with the English corrections. We are grateful to Prof. Rebecca Kimball, Prof. Shou-Hsien Li and Dr. Lu Dong for their comments on the manuscript. We also thank the anonymous reviewers for their comments on the manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of supporting data
The data set supporting the results of this article is available in the NCBI repository. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, A., Chen, D., Wang, P. et al. Characterization of novel microsatellite markers of the Emei Shan Liocichla using restriction site-associated DNA sequencing. Avian Res 8, 13 (2017). https://doi.org/10.1186/s40657-017-0071-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40657-017-0071-8