
Abstract
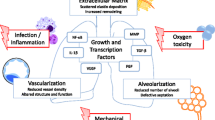
Chronic lung disease of the newborn, also known as bronchopulmonary dysplasia (BPD), is the most common chronic lung disease in early infancy and results in an increased risk for long-lasting pulmonary impairment in the adult. BPD develops upon injury of the immature lung by oxygen toxicity, mechanical ventilation, and infections which trigger sustained inflammatory immune responses and extensive remodeling of the extracellular matrix together with dysregulated growth factor signaling. Histopathologically, BPD is characterized by impaired alveolarization, disrupted vascular development, and saccular wall fibrosis. Here, we explore the hypothesis that development of BPD involves disturbance of conserved pathways of molecular aging that may contribute to premature aging of the lung and an increased susceptibility to chronic lung diseases in adulthood.
Similar content being viewed by others

Introduction
Chronic lung disease of the newborn, also known as bronchopulmonary dysplasia (BPD), is the most common chronic lung disease in early infancy and results in an increased risk for pulmonary and neurologic impairment persisting into adulthood [1]. BPD is defined by the need for supplemental oxygen and/or ventilator support for longer than 28 days, or beyond 36 weeks post-menstrual age, and is classified into three different grades of severity (mild, moderate, severe) [2]. The incidence of BPD is reported up to 77 % in infants born at less than 32 weeks of gestation with a birth weight below 1 kg [3]. Histopathologically, BPD lungs show impaired alveolarization associated with diminished development of small vessels [4, 5]. These structural alterations are accompanied by characteristic inflammatory changes and extensive remodeling of the extracellular matrix (ECM) together with increased smooth muscle mass in small pulmonary arteries and airways [4]. Risk factors for the development of BPD that have been identified by clinical and experimental studies include infections occurring both in utero and post-partum, as well as oxygen toxicity and the impact of mechanical ventilation [6]. Such injuries occur beyond structural and functional immaturity of the organ and the background of genetic susceptibility.
Below, we would like to explore the hypothesis that early lung injury affects conserved pathways of aging thereby contributing to the development of BPD. For that, we first outline the molecular pathways of aging and then summarize available knowledge on how these pathways are affected by experimental hyperoxia and mechanical ventilation of the newborn lung and in BPD. Recent data indicate that adult preterm birth survivors, especially those who developed BPD, exhibit features of clinically relevant respiratory dysfunction later in life [7, 8]. We propose that early alterations in major aging pathways drive premature aging of the lung thereby adding to the risk for development of chronic lung diseases later in life [9, 10].
Review
Molecular concepts of aging
Over the past 30 years, basic and translational research has identified several molecular pathways of aging defined as the “hallmarks of aging,” i.e., genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intercellular communication, and stem cell exhaustion, which provide a molecular foundation for organismal aging [11]. Strikingly, all of these pathways are key pathways for organismal growth, maintenance, and communication. We have recently added an additional hallmark to these molecular pathways, i.e., dysregulation of the ECM, and dissected the distinct aging hallmarks for their differential contribution to the development of age-related chronic lung diseases such as chronic obstructive pulmonary disease (COPD), lung cancer, and idiopathic pulmonary fibrosis (IPF) [12]. Below, we will summarize available evidence that early injury of the neonatal lung as in BPD affects distinct hallmark pathways of aging. This may drive premature aging of the adult lung and early onset of chronic lung diseases in later life.
Molecular pathways of aging are altered by injury of the neonatal lung and in BPD
Hyperoxia as mediated by oxygen supplementation results in increased levels of reactive oxygen species and subsequent oxidative damage of DNA contributing to genomic instability [13, 14]. Very similar to the adult lung, oxygen supplementation of preterm infants induces oxidative stress in the immature lung and causes oxidative modifications of DNA and activation of DNA damage response pathways such as p53 and ATM as observed in hyperoxic ventilated premature baboons and in a rat model of BPD [15–18]. In spontaneous dwarf rats, increased resistance to hyperoxic stress was associated with reduced signs of DNA damage in multiple organs including the lung and contributed to an extended life span of this rat strain compared to their wild-type controls [19]. These data suggest a causal relationship between regulation of DNA damage upon hyperoxia and life span.
Telomere sequences protect the ends of chromosomes and are lost upon DNA replication, thereby limiting the proliferative capacity of cells [20]. Attrition of telomeres over time is a characteristic feature of aging and serves as one of the major markers for premature aging [11]. While there is no evidence for a general association of birth size and telomere length in adult life [21], some recent studies suggest that telomere length in circulating leukocytes or salivary cells is shorter in young adults born preterm compared to that in young adults born at term [22, 23]. Accelerated attrition of telomeres may thus potentially add to the risk of chronic lung diseases as development and progression of both, COPD and IPF, have been associated with reduced telomere function [12].
Development and maturation of the neonatal to the adult lung involves major epigenetic alterations such as changes in DNA methylation, histone marks, and microRNA (miRNA) expression [24–29]. Emerging evidence suggests that early alterations in epigenetic marks are associated with chronic lung diseases such as asthma, COPD, and IPF [30]. Dysregulation of chromatin remodeling pathways including DNA methylation, histone acetylation, and miRNA regulation have also been reported in response to hyperoxia in the neonatal lung in several experimental models and in BPD patients: The process of alveolar septation in the mouse and human lung is accompanied by altered DNA methylation profiles that coincide with distinct changes in gene expression. Of note, dysregulated alveolar septation as observed in BPD was associated with distinct methylation profiles suggesting that abnormal DNA methylation contributes to differential gene expression in human BPD [31]. In particular, bone morphogenetic protein (BMP) 7 showed an inverse correlation between DNA methylation and expression in human BPD samples, with BMP7 being expressed at reduced levels in BPD. As BMP7 opposes the activity of transforming growth factor beta (TGF-β), this may allow enhanced TGF-β signaling in the immature lung contributing pulmonary fibrosis and arrested lung development [32]. Alterations in chromatin remodeling and histone acetylation have also been reported for preterm infants that were at risk for BPD development [33]. In experimental rat and mouse models of neonatal lung injury, hyperoxia was shown to result in diminished expression of histone deacetylases (HDAC) 1 and 2 similar to reduced HDAC activity in lungs of COPD patients but contrary to the increased levels of HDACs in IPF lungs [34–37]. In hyperoxia-treated neonatal rats, DNA methylation by DNMT3b- and EZH2-catalyzed histone methylation was observed [38]. Whether similar DNA and histone modifications also occur in chronic lung diseases remain to be investigated. Pronounced changes in the miRNA profile have also been observed in response to hyperoxia in neonatal lungs during experimental development of BPD [39, 40]. A recent meta-analysis on miRNA profiles in BPD reported upregulation of miRNA-21, miRNA-34a, miRNA-431, and Let-7f and downregulation of miRNA-335 in BPD lung tissues compared to normal groups [41]. Except for miRNA-21, however, there is no major overlap with miRNA profiles of patients with chronic lung diseases [42], suggesting that miRNA regulation in the newborn lung might be inherently different from the adult organ [43]. In summary, these experimental and clinical data suggest that changes in epigenetic programming are associated with neonatal lung damage by oxygen supplementation and BPD development but that there are inherent differences between the immature newborn and adult lung.
Proteostasis, i.e., maintenance of protein homeostasis, ranges from correct protein synthesis, proper protein maturation, folding, and interaction to controlled disposal of unwanted and damaged proteins. Loss of proteostasis has been proposed as a major hallmark of aging [11]. Accumulating data suggest that protein folding and degradation pathways are dysregulated by hyperoxia and mechanical ventilation as recently reviewed by us [44]. For the newborn lung, some evidence suggests that increased stress of the endoplasmic reticulum (ER) [45] and augmented autophagy contributes to hyperoxia-induced surfactant protein (SP)-C accumulation and subsequent injury of neonatal rat lungs [46]. Increased ER stress and autophagy have also been described as characteristic features of IPF and COPD, respectively [47]. Thorough evaluation of protein homeostasis in the preterm lung, however, is missing possibly due to the difficulties in obtaining reliable protein data from small-sized lung samples in experimental and clinical BPD.
Another aging hallmark is deregulated nutrient sensing via the IGF-1/AKT/mTOR axis, an evolutionary conserved growth signaling pathway that integrates nutrient signals to regulate cell growth [48]. For BPD, intrauterine growth restriction that results from nutritional and hormonal (e.g., insulin) deficiencies of the fetus represents an independent risk factor [49, 50]. Insufficient nutrient supply associated with reduced levels of insulin impairs pulmonary alveolar and vessel growth in fetal sheep lungs [51]. On the cellular level, there seems to be a fine-tuned balance of mTOR/Akt activation that needs to be maintained for proper lung maturation: On the one hand, induction of Akt signaling protected neonatal lungs from hypoxia-induced injury [52], while inactivation of the mTOR/Akt pathway negatively regulated SP-A secretion in alveolar epithelial cells and contributed to respiratory distress syndrome in mice or early lethality, respectively [53, 54]. Thus, while the worsening effects of diminished nutrient supply are well established for BPD development, there is only little known about how growth factor signals are integrated via the IGF-1/AKT/mTOR signaling axis and how signaling is regulated in a cell type-specific manner in BPD.
Exhaustion of stem cells has been implicated as a driving factor for several age-related diseases [55, 56]. Most probably, exhaustion of stem cells is not a typical feature of BPD, but impaired mobilization of bone marrow-derived progenitor cells and/or increased sensitivity to oxidative stress might be a contributing factor to BPD disease pathology [57, 58]. Accordingly, reduced recruitment of endothelial progenitor cells (EPC) from the bone marrow has been reported in neonatal mice in response to hyperoxia while adult mice showed rather increased EPC levels [59]. Moreover, reduced numbers of cord vein-derived progenitor cells have been associated with development of BPD [60, 61]. The concept of protective effects of progenitor and stem cells is supported by two recent studies that showed that paracrine effects of exogenously supplemented stem cells alleviate impaired alveolar growth in neonatal lung injury in rats and mice [62, 63]. Therapeutic application of stem cells, e.g., mesenchymal stem cells (MSC), for treatment of BPD is thus a promising option [58, 64, 65].
Mitochondrial dysfunction has been proposed as another hallmark of aging [11]. The free radical theory of aging early on proposed that dysfunctional mitochondria contribute to elevated levels of reactive oxygen species (ROS) and subsequent oxidative damage of the cell [66]. This concept has been extended in recent years to a more complex understanding of mitochondrial metabolism and mitochondrial DNA function in aging [67]. Hyperoxia has been shown to impair both glycolysis and oxidative phosphorylation in alveolar epithelial cells contributing to elevated ROS formation [68]. In the newborn lung, hyperoxia treatment impaired mitochondrial respiration and added to BPD development. Moreover, direct inhibition of oxidative phosphorylation by pyraben, an inhibitor of mitochondrial complex I, resulted in abnormal alveolar development [69]. Another study implicated that only early hyperoxia-induced postnatal mitochondrial ROS production contributes to BPD development [70]. Mitochondrial dysfunction, as, e.g., induced by hyperoxia, may thus contribute to early lung damage of the neonatal lung and development of BPD.
Cellular senescence is a defined cellular program of aging that limits the replicative capacity of cells by cell cycle arrest, thereby preventing propagation of old and damaged cells [71]. Senescence also contributes to tissue remodeling during embryonic development and upon tissue damage [72]. In the neonatal mouse lung, the senescence program was activated by hyperoxia via decreased histone deacetylase activity and upregulation of p21, contributing to impaired alveolarization [34]. The impact of prenatal cigarette smoke on premature senescence may be explained by caveolin-1 expression, linking lung fibroblast senescence and development of pulmonary emphysema [73].
Altered intercellular communication due to disturbed cell-cell signaling is another characteristic feature of aging [11]. The release of cytokines and imbalanced growth factor signaling in the preterm lung, e.g., TGF-β, leads to the activation of different transcription factors and results in a characteristic increase in apoptosis of different cell types [74]. In particular, platelet-derived growth factor-α and the fibroblast growth factor family play a major role in myofibroblast-driven secondary crest formation [75]. The presence of dysmorphic capillaries is related to an altered pattern of angiogenic growth factors such as reduced expression of the vascular endothelial growth factor and its receptors in the neonatal lung [76, 77], accompanied by diminished endothelial nitric-oxide synthase and soluble guanylate cyclase in lung blood vessels and airways [78, 79]. These changes contribute to subsequent development of pulmonary hypertension and impaired lung lymphatic drainage [80]. Perturbation of central signaling pathways, e.g., suppression of the nuclear factor kappa B (NF-kB), disrupts airway-branching and impairs development of epithelial, mesenchymal, and endothelial cell structures culminating in a failure of lung development [81–83]. Moreover, there is altered communication of immune and inflammatory cells with parenchymal cells of the immature lung contributing to innate and adaptive immune responses in BPD [84, 85]. These data clearly indicate that altered intercellular communication as one of the aging hallmarks is of central relevance for the development of BPD and most likely contributes to long-term consequences as the lung matures and ages.
We have recently proposed dysregulation of the ECM as another pillar in the process of lung aging [12]. Similar to disease-relevant processes in the aging adult lung, ECM dysregulation is a characteristic feature of the injured neonatal lung [86]. Experimental studies in rodents and baboons recapitulated ECM alterations and clearly linked ECM remodeling to apoptosis, inflammation, and altered growth factor signaling [85, 87–91]. Accordingly, therapeutic prevention of ECM degradation in the neonatal mouse lung preserved lung growth and structure upon mechanical ventilation [92, 93]. Moreover, the ECM is more than a simple scaffold but also directs the fate of cellular differentiation. This was convincingly shown by several studies which re-populated cells in de-cellularized lungs of different matrix compositions [94–96]. Sustained reorganization of the ECM as observed in BPD may thus not only alter the function of the ECM as a scaffold for lung development but may also affect its potential to regulate cellular differentiation in the lung. As such, dysregulation of the ECM in BPD may contribute to an altered long-term ECM memory that fosters development of chronic lung diseases such as COPD and IPF [12].
Conclusions
We are only beginning to understand the molecular pathways that contribute to the development of BPD. Despite some serious lack of knowledge, there is a remarkable overlap between cellular pathways involved in aging and BPD development, such as genomic instability, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, altered intercellular communication, and ECM remodeling. Regarding the contribution of stem cells, some evidence suggests that instead of stem cell exhaustion, impaired mobilization and recruitment of progenitor cells may contribute to the development of BPD. The contribution of telomere attrition still remains to be investigated. Of note, most of the studies summarized here provide mainly descriptive evidence for an involvement of specific aging pathways in BPD. In order to identify a causal role of distinct aging pathways in the pathogenesis of BPD, one should test the effect of hyperoxia and mechanical ventilation in animal models with genetic modifications of single-pathway components: If these pathways causally contribute to the development of BPD, experimental aggravation would then accelerate damage of the neonatal lung while its experimental amelioration would retard BPD development. The hypothesis that early injury via specific impairment of aging pathways promotes age-related lung damage later in life can also be tested in such experimental models by analyzing lung function over time in adult animals after early injury and upon second hits such as cigarette smoke exposure or infections.
We recently proposed that the essential nature of the hallmarks of aging for the organism makes it very likely that one or the other pathway will be dysregulated in any chronic lung disease [12]. This notion may also apply to the development of BPD. Early dysregulation of cellular and tissue-related maintenance mechanisms such as DNA repair, proteostasis, stem cells, and cell/cell and cell/matrix interactions will then contribute to amplification of cellular damage over time and contribute to impaired lung function later in life. Therapeutic targeting of such central maintenance hubs may thus represent a promising mechanism to interfere with early injury-induced development of chronic lung diseases.
Abbreviations
BPD, bronchopulmonary dysplasia; COPD, chronic obstructive pulmonary disease; ECM, extracellular matrix; IPF, idiopathic pulmonary fibrosis; NF-kB, nuclear factor kappa B; ROS, reactive oxygen species; SP, surfactant protein; TGF-β, transforming growth factor beta
References
Walsh MC, Szefler S, Davis J et al (2006) Summary proceedings from the bronchopulmonary dysplasia group. Pediatrics 117:S52–6. doi:10.1542/peds.2005-0620I
Jobe AH, Bancalari E (2001) Bronchopulmonary dysplasia. Am J Respir Crit Care Med 163:1723–9. doi:10.1164/ajrccm.163.7.2011060
Blencowe H, Cousens S, Chou D et al (2013) Born too soon: the global epidemiology of 15 million preterm births. Reprod Health 10(Suppl 1):S2, 10.1186/1742-4755-10-S1-S2
Coalson JJ (2003) Pathology of new bronchopulmonary dysplasia. Semin Neonatol 8:73–81
Jobe AH (2011) The new bronchopulmonary dysplasia. Curr Opin Pediatr 23:167–72. doi:10.1097/MOP.0b013e3283423e6b
Niedermaier S, Hilgendorff A (2015) Bronchopulmonary dysplasia—an overview about pathophysiologic concepts. Mol Cell Pediatr 2:2. doi:10.1186/s40348-015-0013-7
Caskey S, Gough A, Rowan S, et al. (2016) Structural and functional lung impairment in adult survivors of bronchopulmonary dysplasia. Ann Am Thorac Soc 1–49. doi: 10.1513/AnnalsATS.201509-578OC
Gough A, Linden M, Spence D et al (2014) Impaired lung function and health status in adult survivors of bronchopulmonary dysplasia. Eur Respir J 43:808–16. doi:10.1183/09031936.00039513
Lange P, Marott JL, Vestbo J et al (2012) Prediction of the clinical course of COPD using the new GOLD classification: a study of the general population. Am J Respir Crit Care Med. doi:10.1164/rccm.201207-1299OC
Sly PD, Bush A (2016) From the cradle to the grave: the early-life origins of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 193:1–2. doi:10.1164/rccm.201509-1801ED
López-Otín C, Blasco M, Partridge L et al (2013) The hallmarks of aging. Cell 153:1194–1217. doi:10.1016/j.cell.2013.05.039
Meiners S, Eickelberg O, Konigshoff M (2015) Hallmarks of the ageing lung. Eur Respir J 45:807–827. doi:10.1183/09031936.00186914
O’Reilly MA (2001) DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am J Physiol Lung Cell Mol Physiol 281:L291–305
Barker GF, Manzo ND, Cotich KL et al (2006) DNA damage induced by hyperoxia. Am J Respir Cell Mol Biol 35:277–288. doi:10.1165/rcmb.2005-0340OC
Kulkarni A, Das KC (2008) Differential roles of ATR and ATM in p53, Chk1, and histone H2AX phosphorylation in response to hyperoxia: ATR-dependent ATM activation. Am J Physiol Lung Cell Mol Physiol 294:L998–L1006. doi:10.1152/ajplung.00004.2008
Resseguie E, Staversky RJ, Brookes PS, O’Reilly M (2015) Hyperoxia activates ATM independent from mitochondrial ROS and dysfunction. Redox Biol 5:176–185. doi:10.1016/j.redox.2015.04.012
Das KC, Wasnick JD (2014) Biphasic response of checkpoint control proteins in hyperoxia: exposure to lower levels of oxygen induces genome maintenance genes in experimental baboon BPD. Mol Cell Biochem 395:187–98. doi:10.1007/s11010-014-2124-1
Maniscalco WM, Watkins RH, Roper JM et al (2005) Hyperoxic ventilated premature baboons have increased p53, oxidant DNA damage and decreased VEGF expression. Pediatr Res 58:549–56. doi:10.1203/01.pdr.0000176923.79584.f7
Sasaki T, Tahara S, Shinkai T et al (2013) Lifespan extension in the spontaneous dwarf rat and enhanced resistance to hyperoxia-induced mortality. Exp Gerontol 48:457–463. doi:10.1016/j.exger.2013.02.015
Blackburn EH, Greider CW, Szostak JW (2006) Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 12:1133–8. doi:10.1038/nm1006-1133
Kajantie E, Pietiläinen KH, Wehkalampi K et al (2012) No association between body size at birth and leucocyte telomere length in adult life—evidence from three cohort studies. Int J Epidemiol 41:1400–8. doi:10.1093/ije/dys127
Smeets CCJ, Codd V, Samani NJ, Hokken-Koelega ACS (2015) Leukocyte telomere length in young adults born preterm: support for accelerated biological ageing. PLoS One 10:e0143951. doi:10.1371/journal.pone.0143951
Hadchouel A, Marchand-Martin L, Franco-Montoya M-L et al (2015) Salivary telomere length and lung function in adolescents born very preterm: a prospective multicenter study. PLoS One 10:e0136123. doi:10.1371/journal.pone.0136123
Lui JC, Chen W, Cheung CSF, Baron J (2014) Broad shifts in gene expression during early postnatal life are associated with shifts in histone methylation patterns. PLoS One 9:e86957. doi:10.1371/journal.pone.0086957
Dong J, Carey W, Abel S et al (2012) MicroRNA-mRNA interactions in a murine model of hyperoxia-induced bronchopulmonary dysplasia. BMC Genomics 13:204. doi:10.1186/1471-2164-13-204
Harris KS, Zhang Z, McManus MT et al (2006) Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci U S A 103:2208–13. doi:10.1073/pnas.0510839103
Lui JC, Forcinito P, Chang M et al (2010) Coordinated postnatal down-regulation of multiple growth-promoting genes: evidence for a genetic program limiting organ growth. FASEB J 24:3083–92. doi:10.1096/fj.09-152835
Williams AE, Moschos SA, Perry MM et al (2007) Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev Dyn 236:572–80. doi:10.1002/dvdy.21047
Yuen RK, Neumann SM, Fok AK et al (2011) Extensive epigenetic reprogramming in human somatic tissues between fetus and adult. Epigenetics Chromatin 4:7. doi:10.1186/1756-8935-4-7
Yang IV, Schwartz DA (2011) Epigenetic control of gene expression in the lung. Am J Respir Crit Care Med 183:1295–301. doi:10.1164/rccm.201010-1579PP
Cuna A, Halloran B, Faye-Petersen O et al (2014) Alterations in gene expression and DNA methylation during murine and human lung alveolar septation. Am J Respir Cell Mol Biol 53:60–73. doi:10.1165/rcmb.2014-0160OC
Vicencio AG, Lee CG, Cho SJ et al (2004) Conditional overexpression of bioactive transforming growth factor-beta1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol 31:650–6. doi:10.1165/rcmb.2004-0092OC
Cohen J, Van Marter LJ, Sun Y et al (2007) Perturbation of gene expression of the chromatin remodeling pathway in premature newborns at risk for bronchopulmonary dysplasia. Genome Biol 8:R210. doi:10.1186/gb-2007-8-10-r210
Londhe VA, Sundar IK, Lopez B et al (2011) Hyperoxia impairs alveolar formation and induces senescence through decreased histone deacetylase activity and up-regulation of p21 in neonatal mouse lung. Pediatr Res 69:371–7. doi:10.1203/PDR.0b013e318211c917
Zhu L, Li H, Tang J et al (2012) Hyperoxia arrests alveolar development through suppression of histone deacetylases in neonatal rats. Pediatr Pulmonol 47:264–74. doi:10.1002/ppul.21540
Schamberger AC, Mise N, Meiners S, Eickelberg O (2014) Epigenetic mechanisms in COPD: implications for pathogenesis and drug discovery. Expert Opin Drug Discov 9:609–28. doi:10.1517/17460441.2014.913020
Korfei M, Skwarna S, Henneke I et al (2015) Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 70:1022–32. doi:10.1136/thoraxjnl-2014-206411
Zhu Y, Fu J, Yang H et al (2015) Hyperoxia-induced methylation decreases RUNX3 in a newborn rat model of bronchopulmonary dysplasia. Respir Res 16:75. doi:10.1186/s12931-015-0239-x
Bhaskaran M, Xi D, Wang Y et al (2012) Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol Genomics 44:970–980. doi:10.1152/physiolgenomics.00145.2011
Zhang X, Peng W, Zhang S et al (2011) MicroRNA expression profile in hyperoxia-exposed newborn mice during the development of bronchopulmonary dysplasia. Respir Care 56:1009–15. doi:10.4187/respcare.01032
Yang Y, Qiu J, Kan Q et al (2013) MicroRNA expression profiling studies on bronchopulmonary dysplasia: a systematic review and meta-analysis. Genet Mol Res 12:5195–206. doi:10.4238/2013.October.30.4
Maltby S, Plank M, Tay HL et al (2016) Targeting microRNA function in respiratory diseases: mini-review. Front Physiol 7:1–10. doi:10.3389/fphys.2016.00021
Alvarez-Garcia I, Miska EA (2005) MicroRNA functions in animal development and human disease. Development 132:4653–4662. doi:10.1242/dev.02073
Meiners S, Ballweg K (2014) Proteostasis in pediatric pulmonary pathology. Mol Cell Pediatr 1:11. doi:10.1186/s40348-014-0011-1
Choo-Wing R, Syed M, Harijith A et al (2013) Hyperoxia and interferon-γ-induced injury in developing lungs occur via cyclooxygenase-2 and the endoplasmic reticulum stress-dependent pathway. Am J Respir Cell Mol Biol 48:749–57. doi:10.1165/rcmb.2012-0381OC
Zhang L, Zhao S, Yuan L-J et al (2015) Autophagy regulates hyperoxia-induced intracellular accumulation of surfactant protein C in alveolar type II cells. Mol Cell Biochem 408:181–189. doi:10.1007/s11010-015-2494-z
Meiners S, Keller IE, Semren N, Caniard A (2014) Regulation of the proteasome: evaluating the lung proteasome as a new therapeutic target. Antioxid Redox Signal 21:2364–82. doi:10.1089/ars.2013.5798
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493:338–45. doi:10.1038/nature11861
De Jesus LC, Pappas A, Shankaran S et al (2013) Outcomes of small for gestational age infants born at <27 weeks’ gestation. J Pediatr 163:55–60, 10.1016/j.jpeds.2012.12.097
Zeitlin J, El Ayoubi M, Jarreau P-H et al (2010) Impact of fetal growth restriction on mortality and morbidity in a very preterm birth cohort. J Pediatr 157:733–9.e1. doi:10.1016/j.jpeds.2010.05.002
Rozance PJ, Seedorf GJ, Brown A et al (2011) Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol 301:L860–71. doi:10.1152/ajplung.00197.2011
Park H-S, Park J-W, Kim H-J et al (2013) Sildenafil alleviates bronchopulmonary dysplasia in neonatal rats by activating the hypoxia-inducible factor signaling pathway. Am J Respir Cell Mol Biol 48:105–13. doi:10.1165/rcmb.2012-0043OC
Ikeda H, Shiojima I, Oka T et al (2011) Increased Akt-mTOR signaling in lung epithelium is associated with respiratory distress syndrome in mice. Mol Cell Biol 31:1054–65. doi:10.1128/MCB.00732-10
Miakotina OL, Goss KL, Snyder JM (2002) Insulin utilizes the PI 3-kinase pathway to inhibit SP-A gene expression in lung epithelial cells. Respir Res 3:27
Sánchez Alvarado A, Yamanaka S (2014) Rethinking differentiation: stem cells, regeneration, and plasticity. Cell 157:110–9. doi:10.1016/j.cell.2014.02.041
Behrens A, van Deursen JM, Rudolph KL, Schumacher B (2014) Impact of genomic damage and ageing on stem cell function. Nat Cell Biol 16:201–7. doi:10.1038/ncb2928
Möbius MA, Rüdiger M (2016) Mesenchymal stromal cells in the development and therapy of bronchopulmonary dysplasia. Mol Cell Pediatr 3:18. doi:10.1186/s40348-016-0046-6
Mobius MA, Thébaud B (2015) Stem cells and their mediators—next generation therapy for bronchopulmonary dysplasia. Front Med 2:1–12. doi:10.3389/fmed.2015.00050
Balasubramaniam V, Mervis CF, Maxey AM et al (2007) Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292:L1073–84. doi:10.1152/ajplung.00347.2006
Borghesi A, Massa M, Campanelli R et al (2009) Circulating endothelial progenitor cells in preterm infants with bronchopulmonary dysplasia. Am J Respir Crit Care Med 180:540–6. doi:10.1164/rccm.200812-1949OC
Baker CD, Balasubramaniam V, Mourani PM et al (2012) Cord blood angiogenic progenitor cells are decreased in bronchopulmonary dysplasia. Eur Respir J 40:1516–22. doi:10.1183/09031936.00017312
van Haaften T, Byrne R, Bonnet S et al (2009) Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am J Respir Crit Care Med 180:1131–42. doi:10.1164/rccm.200902-0179OC
Aslam M, Baveja R, Liang OD et al (2009) Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 180:1122–30. doi:10.1164/rccm.200902-0242OC
Chang YS, Ahn SY, Yoo HS et al (2014) Mesenchymal stem cells for bronchopulmonary dysplasia: phase 1 dose-escalation clinical trial. J Pediatr 164:966–972.e6. doi:10.1016/j.jpeds.2013.12.011
O’Reilly M, Thé baud B (2013) The promise of stem cells in bronchopulmonary dysplasia. Semin Perinatol 37:79–84. doi:10.1053/j.semperi.2013.01.003
Harman D (1981) The aging process. Proc Natl Acad Sci U S A 78:7124–8
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120:483–95. doi:10.1016/j.cell.2005.02.001
Das KC (2013) Hyperoxia decreases glycolytic capacity, glycolytic reserve and oxidative phosphorylation in MLE-12 cells and inhibits complex I and II function, but not complex IV in isolated mouse lung mitochondria. PLoS One 8:e73358. doi:10.1371/journal.pone.0073358
Ratner V, Starkov A, Matsiukevich D et al (2009) Mitochondrial dysfunction contributes to alveolar developmental arrest in hyperoxia-exposed mice. Am J Respir Cell Mol Biol 40:511–8. doi:10.1165/rcmb.2008-0341RC
Datta A, Kim G, Taylor JM et al (2015) Mouse lung development and NOX1 induction during hyperoxia are developmentally regulated and mitochondrial ROS-dependent. Am J Physiol - Lung Cell Mol Physiol. doi:10.1152/ajplung.00176.2014
Rodier F, Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192:547–56. doi:10.1083/jcb.201009094
Muñoz-Espín D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15:482–96. doi:10.1038/nrm3823
Volonte D, Galbiati F (2009) Caveolin-1, cellular senescence and pulmonary emphysema. Aging (Albany NY) 1:831–5
Kunzmann S, Speer CP, Jobe AH, Kramer BW (2006) Antenatal inflammation induced TGF-beta1 but suppressed CTGF in preterm lungs. AJP Lung Cell Mol Physiol 292:L223–L231. doi:10.1152/ajplung.00159.2006
Jankov RP, Keith Tanswell A (2004) Growth factors, postnatal lung growth and bronchopulmonary dysplasia. Paediatr Respir Rev 5:S265–S275. doi:10.1016/S1526-0542(04)90050-4
De Paepe ME, Mao Q, Powell J et al (2006) Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med 173:204–211. doi:10.1164/rccm.200506-927OC
Thebaud B (2007) Angiogenesis in lung development, injury and repair: implications for chronic lung disease of prematurity. Neonatology 91:291–297. doi:10.1159/000101344
Vyas-Read S, Shaul PW, Yuhanna IS, Willis BC (2007) Nitric oxide attenuates epithelial-mesenchymal transition in alveolar epithelial cells. AJP Lung Cell Mol Physiol 293:L212–L221. doi:10.1152/ajplung.00475.2006
Bland RD, Ling CY, Albertine KH et al (2003) Pulmonary vascular dysfunction in preterm lambs with chronic lung disease. Am J Physiol Lung Cell Mol Physiol 285:L76–85. doi:10.1152/ajplung.00395.2002
Kinsella JP, Greenough A, Abman SH (2006) Bronchopulmonary dysplasia. Lancet 367:1421–1431. doi:10.1016/S0140-6736(06)68615-7
Yang G, Abate A, George AG et al (2004) Maturational differences in lung NF-kB activation and their role in tolerance to hyperoxia. J Clin Invest 114:669–678. doi:10.1172/JCI200419300
Alvira CM, Abate A, Yang G et al (2007) Nuclear factor-kappaB activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am J Respir Crit Care Med 175:805–15. doi:10.1164/rccm.200608-1162OC
Iosef C, Alastalo T-P, Hou Y et al (2012) Inhibiting NF- B in the developing lung disrupts angiogenesis and alveolarization. AJP Lung Cell Mol Physiol 302:L1023–L1036. doi:10.1152/ajplung.00230.2011
Balany J, Bhandari V (2015) Understanding the impact of infection, inflammation, and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med 2:90. doi:10.3389/fmed.2015.00090
Kompass KS, Deslee G, Moore C et al (2010) Highly conserved transcriptional responses to mechanical ventilation of the lung. Physiol Genomics 42:384–396. doi:10.1152/physiolgenomics.00117.2009
Hilgendorff A, O’Reilly MA (2015) Bronchopulmonary dysplasia early changes leading to long-term consequences. Front Med 2:1–10. doi:10.3389/fmed.2015.00002
Bland RD, Ertsey R, Mokres LM et al (2008) Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol 294:L3–14. doi:10.1152/ajplung.00362.2007
Mokres LM, Parai K, Hilgendorff A et al (2010) Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. AJP Lung Cell Mol Physiol 298:L23–L35. doi:10.1152/ajplung.00251.2009
Kroon AA, Wang J, Kavanagh B et al (2011) Prolonged mechanical ventilation induces cell cycle arrest in newborn rat lung. PLoS One 6:e16910. doi:10.1371/journal.pone.0016910
Yasumatsu R, Altiok O, Benarafa C et al (2006) SERPINB1 upregulation is associated with in vivo complex formation with neutrophil elastase and cathepsin G in a baboon model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 291:L619–27. doi:10.1152/ajplung.00507.2005
Altiok O, Yasumatsu R, Bingol-Karakoc G et al (2006) Imbalance between cysteine proteases and inhibitors in a baboon model of bronchopulmonary dysplasia. Am J Respir Crit Care Med 173:318–26. doi:10.1164/rccm.200503-425OC
Hilgendorff A, Parai K, Ertsey R et al (2011) Inhibiting lung elastase activity enables lung growth in mechanically ventilated newborn mice. Am J Respir Crit Care Med 184:537–546. doi:10.1164/rccm.201012-2010OC
Hilgendorff A, Parai K, Ertsey R et al (2012) Neonatal mice genetically modified to express the elastase inhibitor elafin are protected against the adverse effects of mechanical ventilation on lung growth. AJP Lung Cell Mol Physiol 303:L215–L227. doi:10.1152/ajplung.00405.2011
Parker MW, Rossi D, Peterson M et al (2014) Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J Clin Invest 124:1622–35. doi:10.1172/JCI71386
Jensen T, Roszell B, Zang F et al (2012) A rapid lung de-cellularization protocol supports embryonic stem cell differentiation in vitro and following implantation. Tissue Eng Part C Methods 18:632–646. doi:10.1089/ten.tec.2011.0584
Bonvillain RW, Danchuk S, Sullivan DE et al (2012) A nonhuman primate model of lung regeneration: detergent-mediated decellularization and initial in vitro recellularization with mesenchymal stem cells. Tissue Eng Part A 18:2437–2452. doi:10.1089/ten.tea.2011.0594
Authors’ contributions
SM prepared the final manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Meiners, S., Hilgendorff, A. Early injury of the neonatal lung contributes to premature lung aging: a hypothesis. Mol Cell Pediatr 3, 24 (2016). https://doi.org/10.1186/s40348-016-0052-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40348-016-0052-8