
Abstract
Background
Roots are the primary site for plant-microbe interactions. Among the three root-associated layers (i.e., rhizosphere, rhizoplane, and endorhiza), the rhizoplane is a key component serving a critical gating role that controls microbial entry into plant roots. The microbial communities colonizing the three layers are believed to be gradually enriched from the bulk soil inoculum. However, it is unknown how this enrichment process, particularly the rhizosphere to rhizoplane step, is affected by biotic stresses, such as disease. In this study, we address this question using the citrus root-associated microbiome as a model.
Results
We identified the rhizosphere-to-rhizoplane-enriched taxonomic and functional properties of the citrus root-associated microbiome and determined how they were affected by Huanglongbing (HLB), a severe systemic disease caused by Candidatus Liberibacter asiaticus, using metagenomic and metatranscriptomic approaches. Multiple rhizoplane-enriched genera were identified, with Bradyrhizobium and Burkholderia being the most dominant. Plant-derived carbon sources are an important driving force for the enrichment process. The enrichment of functional attributes, such as motility, chemotaxis, secretion systems, and lipopolysaccharide (LPS) synthesis, demonstrated more active microbe-plant interactions on the rhizoplane than the rhizosphere. We observed that HLB impaired the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome in three ways: (1) by decreasing the relative abundance of most rhizoplane-enriched genera; (2) by reducing the relative abundance and/or expression activity of the functional attributes involved in microbe-plant interactions; and (3) by recruiting more functional features involved in autotrophic life cycle adaptation, such as carbon fixation and nitrogen nitrification in the HLB rhizoplane microbiome. Finally, our data showed that inoculation of Burkholderia strains isolated from the healthy citrus root-associated microbiome could trigger the expression of genes involved in induced systemic resistance in inoculated plants.
Conclusions
HLB causes decreased relative abundance and/or expression activity of rhizoplane-enriched taxonomic and functional properties, collectively resulting in impaired plant host-microbiome interactions. Manipulation of the citrus root-associated microbiome, for instance, by inoculating citrus roots with beneficial Burkholderia strains, has potential to promote plant health. Our results provide novel insights for understanding the contributions of the community enrichment process of the root-associated microbiome to the plant hosts.
Similar content being viewed by others


Background
Plants harbor a high diversity of microorganisms proximal to, on, and inside their tissues. These microorganisms, which mostly include bacteria, Archaea, and fungi, form microbial communities that are associated with various plant habitats, including the rhizosphere, rhizoplane, phyllosphere, and endosphere, and the communities are collectively known as plant microbiomes [1,2,3]. The plant microbiome can determine the fate of plants in multiple ways, including (i) supplying plants with nutrition, e.g., nitrogen fixation or phosphate solubilization; (ii) modulating plant growth or relieving stress through phytohormone production or degradation; (iii) maintaining plant health through competition with pathogens or induction of plant resistance [4,5,6,7]; and (iv) driving the evolution of multi-disease resistance during long-term coevolution history [8]. Given its importance, the microbiome is considered an integral component of the plant-host assemblage, which is recognized as a “holobiont” [4]. Understanding the plant microbiome will have important implications for the human food supply and its security, biodiversity, and ecosystem functionality [9, 10].
The plant microbiome is predominantly assembled from external inoculum pools, including soil- and air-borne microbial communities [6]. Soil is the largest known reservoir of microbial diversity [11], and roots are the primary site for plant-microbe interactions. Recently, several studies were performed to reveal the assembly mechanisms of the plant root-associated microbiome [12,13,14,15,16,17]. Based on these studies, a three-step enrichment model is proposed for the root-associated microbiome assembly process. Briefly, a fraction of microorganisms in the bulk soil inoculum pool are enriched toward the roots by the general gradients of carbon source, phytochemicals, pH, oxygen, and nutrients imposed by the roots, and these form the rhizosphere microbiome. Secondly, a more specialized community is further enriched on the rhizoplane; more intimate microbe-host interactions occur and much stronger selection pressure from the plant host is applied at the assembled rhizoplane microbiome than the rhizosphere microbiome. Finally, certain microorganisms enter and inhabit the endorhiza, and the established endorhiza microbiome contributes significantly to plant fitness, including immune system modulation [5]. Therefore, among the three root-associated layers described above, the rhizoplane is a key component serving a critical gating role that controls microbial entry into plant roots. However, these previous studies were mainly dedicated to revealing the effects of the plant genomic background on the microbial community assembly process [12,13,14,15,16,17]; how the enrichment process of the microbiome is affected by biotic stresses such as disease remains elusive. In addition, the studies were conducted using annual plants including Arabidopsis, soybean, barley, wheat, cucumber, and rice. Compared to the short lifespan of annual plants, perennial plants are subjected to longer periods of plant-microbe interactions for a single generation, including complicated root growth patterns and variable environmental factors. Compared to annual plants, the microbiome assembly cues of perennial plants are less studied. Here, we investigated the microbial community assembly process of the root-associated microbiome of citrus, a perennial plant, and how Huanglongbing (HLB) affects the microbiome enrichment process.
HLB is the most devastating citrus disease worldwide [18, 19]. The disease is caused by the gram-negative, phloem-limited, alpha-proteobacteria Candidatus Liberibacter spp., i.e., Ca. L. asiaticus (Las), Ca. L. africanus (Laf), and Ca. L. americanus (Lam) [18, 19]. HLB impairs phloem transportation of photoassimilates [18] and causes root decline [20]. Las living in the phloem prevents direct interactions with other microbes on the rhizoplane and in the rhizosphere. Instead, Las causes decreased photoassimilate transportation, likely reducing plant resource availability for the root-associated microbiome. Our previous results suggested that HLB significantly altered the structure or functional potential of the citrus endosphere (leaves or roots) or rhizosphere bacterial community based on cultivation, 16S rDNA clone library, PhyloChip, or the GeoChip method [21,22,23,24]. However, the low-throughput-based methods used in these previous studies limited our understanding of the citrus microbiome. Additionally, these studies focused on the bacterial community colonized on a single layer, either the rhizosphere or endosphere. How HLB affects the microbiome assembly process, particularly the process from the less tightly associated rhizosphere to the rhizoplane component, of the citrus root-associated microbiome remains unknown. In this study, we obtained snapshots of the citrus root microbiome using metagenomic (MG) and metatranscriptomic (MT) approaches to investigate the rhizosphere-to-rhizoplane-enriched taxa and functions and how HLB affects those enriched taxonomic and functional attributes.
Methods
Sample collection
Three healthy and three Las-infected 11-year-old “midsweet” sweet orange on Swingle citrumelo rootstock from a grove at Auburndale (28.11 N, 81.79 W), Florida, USA were identified based on visual symptoms and qPCR results [24] from both leaf and root samples (Additional file 1: Figure S1). The fine root cores (i.e., roots with approximately 2-cm-thick adjacent soil layers) from four corners of each tree were collected for DNA and RNA extraction. The loosely attached soil on the roots was removed with gentle shaking. The rhizosphere soil was carefully and quickly collected by gently brushing the remaining soil adhering to the roots using brush pencils. The soil collection step was performed on ice. Then, the roots were placed in pre-cooled PBS (phosphate-buffered saline) buffer, and the rhizoplane soil was extracted by ultra-sonication as described by Edwards et al. [17]. The roots were sonicated twice for 20 s each (time interval 5 s) using a sonication bath (power 130 W, 60 Hz, Fisher Scientific). The roots were discarded, and the rhizoplane soil was collected by centrifugation at 12,000×g for 1 min at 4 °C. Each rhizosphere or rhizoplane soil sample was re-suspended in LifeGuard soil preservation solution (Mobio Laboratories) immediately after processing. The full procedure was performed sample by sample and as quickly as possible (three persons worked together, total processing time was <5 min for each sample). The soil samples from the four corners of each tree were pooled into a single sample. The samples were stored at −20 °C until further processing.
Nucleic acid extraction and sequencing
DNA and RNA were extracted from 2 g of each soil sample using a RNA PowerSoil total RNA isolation kit and the RNA PowerSoil DNA Elution Accessory Kit following the manufacturer’s protocol (MO BIO Laboratories, Inc.). In this extraction process, DNA and RNA were extracted from the same sample simultaneously (i.e., DNA and RNA were eluted from the same column with different elution buffer), and thus, the RNA samples reflect the real-time gene expression profiling of the corresponding DNA samples. Large-scale shotgun metagenome and metatranscriptome sequencing were performed on the Illumina Hiseq4000 platform by Novogene (Novogene, Beijing, China). Briefly, the DNA samples were randomly sheared using Covaris Ultrasonic Processor into approximately 300 bp fragments, which were then used to construct the sequencing libraries using the Illumina TruSeq® DNA PCR-free sample preparation kit (Illumina, USA). For the RNA samples, the ribosomal RNA was depleted using the Ribo-Zero™ rRNA Removal Kit for Bacteria (Illumina, Madison, USA) according to the manufacturer’s instructions. The remaining transcripts were fragmented and reverse-transcribed. The messenger RNA (mRNA) libraries were prepared using the TrueSeq Stranded mRNA Sample Prep kit (Illumina, USA), and 2 × 125-bp paired-end reads were generated for all samples. Approximately 10 Gb sequencing data for each DNA shotgun sequencing sample and 8 Gb data for each RNA sample were obtained. The DNA and RNA reads were deposited at NCBI under the bioproject accession no. PRJNA324090 and SRA accession no. SRP076109.
Bioinformatics analyses
The raw reads from metagenome sequencing were filtered, trimmed, and quality-controlled to generate the clean reads, which were further trimmed using Sickle [25] with the parameters –q 20 and –l 80. On average, 4.85% of the clean reads were discarded from this trimming step. The trimmed reads were aligned to the Swingle citrumelo genome [26], sweet orange genome [27], and Citrus clementina genome [28] using bowtie2 [29], and the corresponding mapped reads were removed. Only the reads that did not map to any of the three citrus genomes were retained for further analysis.
The filtered reads from all 12 samples were pooled and subjected to de novo assembly using megahit v1.03 [30]. The metagenes were predicted using MetaGeneMark [31]. The non-redundant gene categories (unigenes) were generated using CD-HIT-est with an identity cutoff of 95% [32]. To obtain the taxonomic annotation for the unigenes, the protein sequences were aligned against the NCBI microbial NR database using DIAMOND software [33] with an E value cutoff of 1e-5. Then, the taxonomic annotations were assigned using the MEGAN LCA annotation method [34]. The functional annotation was assigned to the unigenes by blasting against the KEGG orthology database using DIAMOND software.
To generate taxonomic and functional abundance and expression profiling, the short DNA and RNA reads from each sample were aligned to the unigenes using SOAP2 [35] with default parameters. The generated alignments were parsed, and abundance and expression profiling were obtained (reads count matrixes). Based on the abundance and expression profiles, the features (genera and KOs) with significantly differential abundance or expression activity were determined using DESeq2 with a negative binomial generalized linear model (p < 0.05) [36]. For the rhizoplane-enriched feature detection, paired DESeq2 comparison analysis was performed separately for healthy samples and HLB samples to prevent the potential effects of health status on the results, and the identified features were merged. Other comparisons, such as healthy vs. HLB samples for differential rhizoplane taxonomic and functional feature determination, were conducted using DESeq2, where the individual samples from the healthy or HLB trees were treated as biological replicates. The taxonomic and functional dissimilarity analyses among samples were performed using the R package VEGAN [37] with a Bray-Curtis distance matrix. The variation partitioning analysis (VPA) was performed based on the taxonomic and functional composition matrix calculated on the genus and KEGG orthologue (KO) level of all 12 samples (rhizosphere and rhizoplane, healthy and HLB) using VEGAN. The heatmap and Venn diagram plots were drawn using the gplots and VennDiagram package [38, 39], respectively. The average genome size (AGS) for the metagenomic samples was estimated using MicrobeCensus v1.0.7 based on 10 million reads [40]. The bacterial secretion system-based effector genes were identified by blasting the unigenes against a custom-built type III, IV, and VI effector library (T3SE: http://effectors.bic.nus.edu.sg/T3SEdb/index.php; T4SE: http://sate.cirad.fr/; T6SE: http://db-mml.sjtu.edu.cn/SecReT6/) with an amino acid identity cutoff of 45% and a coverage of 80% [41]. To explore the relative contribution of taxa to the rhizosphere to rhizoplane-enriched KOs, the taxonomic information for each selected gene (at genus and family level) was extracted, and their relationships were calculated using the method described by Ofek-Lalzar et al. [15]. The metagenome-assembled genome (MAG) extraction was performed using MetaBAT [42], and the quality of the generated MAGs was checked using CheckM [43]. The genome similarity between selected MAGs and their related genomes was determined using the GGDC2.1 server [44]. The MAGs were annotated using the RAST server and blast2GO software [45, 46]. The annotated bin.74 and bin.105 were deposited in the RAST server with the job id 311713 and 312590, respectively.
Burkholderia sp. isolation and survival assay
Bacterial strains were isolated from the rhizosphere of healthy citrus plants in a relevant study (Riera et al., Unpublished). Based on the 16S rDNA gene sequencing results using the universal primers 27F and 1492R [47], the strains were identified. Burkholderia strains were selected for the antagonistic activity test against several known citrus pathogens. Two representative Burkholderia strains, namely, Burkholderia metallica (strain A53) and Burkholderia territori (strain A63), which showed the best antagonistic activities among the tested strains, were selected for further analysis.
The two strains were transformed with the pUFR034-gfp plasmid by electroporation. Positive GFP (green fluorescent protein)-labeled strains were verified by fluorescent microscopy and qPCR results. Each transformed Burkholderia strain was applied by soil drench (15 mL of 1 × 108 cfu/mL) to five 1-year-old potted “Valencia” sweet orange plants grown in a quarantine greenhouse facility at the Citrus Research and Education Center, University of Florida, in Florida, USA, with controlled temperature (28–35 °C) and a relative humidity of 80%. The rhizosphere and rhizoplane soil samples were collected as described above (but without the LifeGuard soil preservation solution resuspension step) 1 h after inoculation (0 days post inoculation (dpi)), 2, 5, and 9 dpi. The genomic DNA was extracted using a PowerSoil DNA Isolation Kit (MO BIO Laboratories, Inc.). The population dynamics of the inoculated strains inside the rhizosphere and rhizoplane bacterial community (calculated as relative abundance change compared to the 0 dpi data) were calculated [48], with the total bacteria population serving as the reference. A primer pair targeting the GFP gene (GFPF 5′-TCCATGCCATGTGTAATCCC-3′, GFPR 5′-CCATTACCTGTCCACACAATCT-3′) was used for the detection of the inoculated strains. qPCR assays were conducted on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using a Quantifast® SYBER® Green PCR kit (QIAGEN) following the manufacturer’s instructions with the following cycling conditions: an initial denaturation step of 5 min at 95 °C and 35 cycles of 10 s at 95 °C and 30 s at 60 °C. The primer set Eub338 and Eub518, which targets the conserved region of the 16S rRNA gene, was used for the detection of total bacteria, and qPCR assays were performed as described by Fierer et al. [49]. Furthermore, the long-term survival rate of the strains was also determined using 2-year-old Duncan grapefruit plants with three plants/replicates grown under the same growth conditions described above. One gram of rhizosphere soil from each tree was collected, and colony-forming units (CFU) were calculated using nutrient agar plates supplemented with 50 μg/ml kanamycin (strain native resistance for both strains). The CFU formation was determined at 0, 46, 60, 72, 91, 110, 131, 150, and 224 dpi.
Expression of plant defense-related genes in response to beneficial bacteria treatment
Fifteen milliliters of 1 × 108 cfu/mL of strains A53 and A63 were inoculated onto 1-year-old Valencia plants. Application of 15 mL of acibenzolar-S-methyl (ASM) (active ingredient in Actigard 50WG) at 0.33 mg/ml was used as a positive control. The active ingredient of Actigard is an analogue of salicylic acid and systemic elicitor of plant defense [50]. A water only inoculation was used as a negative control. For each treatment, nine seedlings were used, with each seedling as a replicate.
To determine whether the treatments induced plant defense, we measured the expression of three defense-related genes: SAM, encoding S-adenosyl-l-methionine-salicylic acid carboxyl methyltransferase; PR1, encoding pathogenesis-related protein 1; and PR2, encoding pathogenesis-related protein 2 [50] using quantitative reverse transcription PCR (qRT-PCR). The primer sequences for SAM and PR1 are SAMF 5′-GGACGCATCTTCTTGGGATAA-3′/SAMR 5′-CGTGACAGTTTCCTTGACGA-3′ and PR1F 3′-CAGGGTCTCCAAGCAACTATG-5′/PR1R 5′-CCACCTCGCGTATTTCTCTAA-3′, respectively. The primer sequences for PR2 were described previously [50]. Gene expression was determined at 0, 3, 5, and 7 dpi. For each time point, three biological repeats were sampled by collecting one leaf from three different plants. RNA was extracted using an RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. Samples were treated with Ambion® DNA-free DNase Treatment and Removal Reagents. The qRT-PCR was performed using a Verso 1-step RT-qPCR Kit (ThermoFisher), and the fold change was calculated using the ∆∆Ct method as previously described [48].
Results
Structure and function of citrus root-associated microbiome
We collected the rhizosphere and rhizoplane soil samples from three healthy and three HLB-diseased citrus trees for MG and MT analyses. We generated 515,129,383 paired-end clean reads, which is equal to 129 Gb for MG, and 413,225,469 paired-end clean reads, which is equal to 103 Gb for MT, for the 12 samples. The citrus host-originated MG reads were depleted by aligning the reads to the three available citrus draft genomes to comprehensively remove the host-originated reads, and 0.039 to 1.79% clean reads were removed (Additional file 1: Table S1). We pooled the MG reads from all 12 samples in total of 501,171,627 paired-end reads (approximately 120 Gb) for de novo assembly and obtained an assembly of 10.84 Gb across 17,676,569 contigs, with the longest contig at 536,098 bp and N50 at 651 bp (based on all contigs ≥200 bp) (Additional file 1: Table S2). In total, 22,192,564 putative protein-coding genes were predicted from the assembly. After removing redundant sequences (identity >95%), 21,380,400 unigenes were generated. The unigenes represented more rhizoplane reads (46.48 ± 4.98%, mean ± SD, same herein) than rhizosphere reads (35.44 ± 2.34%) (p = 0.0006).
Taxonomy annotation was successfully assigned to 70% of the total unigenes. Bacteria comprised the predominant domain (99.30 ± 0.37%, mean relative abundance ± SD, n = 12), with small fractions of Archaea, eukaryotes, and viruses detected based on the annotated unigenes. Proteobacteria (74.56 ± 8.59%), Actinobacteria (16.80 ± 6.95%), Bacteroidetes (2.86 ± 0.97%), and Acidobacteria (2.42 ± 0.69%) were the dominant phyla (relative abundance ≥1%) (Additional file 1: Figure S2). Thaumarchaeota was the dominant phylum in the Archaea domain (0.41 ± 0.31%), and Ascomycota represented the most abundant phylum affiliated with fungi (0.14 ± 0.06%). Using Diamond BLASTP against the KEGG KO database, 53% of the unigenes were assigned KO function annotation, with most of the KO-annotated genes (94.3%) mapped to the KEGG pathways. In total, 8816 KOs were identified from the unigenes. KOs involved in amino acid metabolism (9.56 ± 0.06%), carbohydrate metabolism (8.65 ± 0.09%), membrane transport (8.51 ± 0.37%), and energy metabolism (5.67 ± 0.10%) were dominant (relative abundance ≥5%) based on KEGG level 2 pathway annotations.
Approximately 3.05 ± 1.39% of the metatranscriptomic reads were identified as rRNA, as derived by the SortMeRNA ver. 2.1 program [51], indicating a high-efficiency rRNA depletion strategy during the MT library construction. In addition, 32.94 ± 8.36% of the RNA clean reads from the rhizoplane samples and 27.38 ± 4.91% from the rhizosphere samples could be mapped to the unigenes. On average, 24.26% (ranging from 18.01 to 41.14%) of the unigenes identified in each sample were actively expressed (Additional file 1: Table S3). The community composition results from the MG and MT analyses revealed a similar structure when tested at the genus level (correlation coefficient r = 0.90, p < 0.005).
Effects of HLB on the rhizosphere- to rhizoplane-enriched taxa
The alpha diversity (Shannon index) of the rhizosphere samples was significantly increased compared to the rhizoplane samples at low (e.g., phylum) or high (e.g., genus) resolution (Additional file 1: Table S4). The AGS of the rhizoplane microbiome was slightly but significantly increased compared to the rhizosphere microbiome (6.31 ± 0.52 Mb for rhizoplane and 5.87 ± 0.26 Mb for rhizosphere, mean ± SD, n = 6, paired t test, p = 0.01). However, no significant difference in alpha diversity or AGS due to HLB was observed for the rhizosphere and rhizoplane samples. We also performed a variation partitioning analysis to reveal the contribution of the sample layer (rhizosphere/rhizoplane) and health status (healthy/HLB) to the observed taxonomic composition difference between samples. The sample layer was the major determinant of the citrus root-associated microbiome (accounting for 78.87% of the variation observed in the entire microbial community, p = 0.024), whereas the contribution of health status was minor (9.32%, p = 0.501). The results collectively suggested that the rhizoplane microbiome harbored a distinct, less complex microbial community, in which microorganisms with larger genome size were more abundant than the rhizosphere microbiome. In addition, HLB did not significantly alter the overall structure of the root-associated microbiome (Additional file 1: Figure S2).
Owing to the intimate relationship between microbes and hosts in the rhizoplane, certain microbes are enriched from rhizosphere-to-rhizoplane, as driven by plant selection [5]. The relative abundance of Proteobacteria was significantly higher in the rhizoplane samples than the rhizosphere samples, and Actinobacteria and Acidobacteria were depleted from the rhizosphere to rhizoplane samples (Additional file 1: Figure S3). A more detailed pairwise comparison at the genus level between the rhizosphere and rhizoplane microbial community was performed. Among the 1950 genera identified in all samples, 119 genera exhibited significantly increased relative abundance in the rhizoplane microbiome compared to the rhizosphere microbiome. Among them, Bradyrhizobium and Burkholderia whose genome sizes are 8.57 ± 0.87 Mb (n = 8) and 7.15 ± 0.84 Mb (n = 124) based on the available complete genomes in MBGD, respectively (accessed September 13, 2016) [52], were ranked as the top two dominant genera. The average relative abundance of Bradyrhizobium and Burkholderia were 50.10 ± 5.22% (mean ± SD, n = 6) and 10.36 ± 4.60% for the rhizoplane samples and 36.96 ± 6.07% and 5.66 ± 3.99% for the rhizosphere samples, respectively. A positive correlation between AGS and the relative abundance of Bradyrhizobium was also observed (Spearman’s rank-order correlation, rs = 0.49, p < 0.1, n = 12). No obvious correlation was observed for Burkholderia and AGS, possibly because of its relative low abundance in the samples. Given the large genome size and significantly increased relative abundance in the rhizoplane microbiome compared to the rhizosphere microbiome, we reasoned that Bradyrhizobium was the main contributor of the increased AGS observed in the rhizoplane microbiome.
Fifty-two of the 119 rhizoplane-enriched genera exhibited significantly different relative abundance between healthy and HLB rhizoplane samples. Notably, 50 of these 52 genera were more abundant in healthy rhizoplane samples than HLB samples (Fig. 1 and Additional file 1: Figure S4), and the other two genera, Inquilinus and Aureimonas, contain known human pathogens [53, 54] and exhibited increased relative abundance in HLB rhizoplane samples compared to healthy samples. Among these 50 genera, Bradyrhizobium and Burkholderia [55, 56] together with several other known root-associated microorganisms such as Variovorax [57], Bdellovibrio [58], Chryseobacterium [59, 60], Dyadobacter [61], and Penicillium from the fungi kingdom [62] were observed. Cellvibrio, which is rhizoplane-enriched and has plant growth-promoting capacity [15, 63], exhibited significantly increased relative abundance in healthy rhizoplane samples compared in HLB samples (Additional file 1: Figure S5A). A nearly complete genome (genome size 5.7 Mb, completeness 99.14% and contamination 0.23% by checkM [43], composing of 41 contigs) representing a novel species of Cellvibrio (Additional file 1: Table S5) was generated using the metagenome-assembled genome (MAG) extraction approach [42]. Plant cell wall polysaccharide-degrading enzymes are key features in rhizoplane microbiome [15]. In total, 350 of 4794 genes in the assembled Cellvibrio bin were involved in carbohydrate utilization as suggested by dbCAN [64], which was significantly increased compared to the non-plant-associated relatives, such as Escherichia coli K12 (132 of 4372 genes are identified, Fisher’s exact test, p < 0.0001). Multiple plant growth-promoting-associated genes, such as genes involved in IAA synthesis, were present and actively expressed (Additional file 1: Figure S5B).

Top rhizoplane-enriched genera with different relative abundances between healthy and HLB rhizoplane samples (displayed by pie chart). The average relative abundance of each taxon in the rhizoplane samples is displayed as a percentage. HLB.P (blue): rhizoplane samples from HLB trees; H.P (red): rhizoplane samples from healthy trees. The genome size for each genus was calculated as the average genome size based on the available complete genomes from MBGD. The full list is shown in Additional file 1: Figure S4
Ninety-four rhizoplane-depleted genera were identified, which were mainly affiliated with Actinobacteria, Acidobacteria, and Archaea, including Thaumarchaeota. The majority of rhizoplane-depleted taxa with significantly different relative abundance between healthy and HLB samples (36 of 39 genera) exhibited significantly increased relative abundance in the HLB rhizoplane microbiome compared to healthy samples (Additional file 1: Figure S6). Thaumarchaea represents an autotrophic microorganism in the soil microbial community [65]. The relative abundance of Thaumarchaea significantly decreased from the rhizosphere to the rhizoplane (p < 0.05) and exhibited significantly increased relative abundance in the rhizoplane microbiome of HLB plants compared to healthy trees (Additional file 1: Figure S7A). One high-quality Thaumarchaeota MAG was extracted from the metagenome assembly (3.17 Mb, completeness 93.2%, and contamination rate 0.97%). The amoA gene-based phylogenetic tree demonstrated that the Thaumarchaeota MAG (bin.105) was affiliated with the Thaumarchaeota group I.1b (Additional file 1: Figure S7B). The low similarity between bin.105 and the other three available Thaumarchaeota group I.1b genomes demonstrated that bin.105 represents a novel member of the Thaumarchaeota phylum (Additional file 1: Table S6). Multiple genes involved in the autotrophic life cycle, including nitrification-associated genes (including amoA and amoB, urease-encoding genes ureABCEFGD) and carbon fixation-associated genes (including 4-hydroxybutyrate-CoA dehydratase, acetyl-CoA carboxylase, and methylmalonyl-CoA), are present in bin.105.
Effects of HLB on rhizosphere-to-rhizoplane-enriched functional properties
Both the KO presence/absence and relative abundance profiling demonstrated that all samples shared very similar functional attributes (Fig. 2a, b). In total, 89.7% of the identified KOs (7938 of 8816) were found in all samples, and the composition similarity among these samples was greater than 98.7%, suggesting that the functional composition of the citrus root-associated microbiome was more conserved than the taxonomic composition (p < 2.2E−16) (Fig. 2a, b and Additional file 1: Figure S2). The sample layer contributed significantly to the functional composition variation (80.84%, p = 0.036) with a minute contribution from health status (9.57%, p = 0.497). We performed the layer KO enrichment analysis to identify the rhizoplane-enriched functional properties. We then determined how HLB affected these rhizoplane-enriched properties at both the genetic potential and gene expression levels. In total, 2218 rhizoplane-enriched KOs were identified. The KOs belonging to the transporter, two-component system, ABC transporter, secretion system, transcription factors, and bacterial motility proteins were the top six enriched functional attributes, highlighting the importance of these functions for microorganisms to adapt to the plant-root surface niche. All identified rhizoplane-enriched KOs were actively expressed, as revealed by the metatranscriptomic data (Additional file 2).

Functional composition of citrus root-associated microbiome. a Venn diagram depicting number of KOs identified in rhizosphere and rhizoplane microbiomes from healthy and HLB-diseased samples. b Heatmap exhibiting functional composition similarity between samples based on relative abundance data at KO level. c Rhizosphere- to rhizoplane-enriched and depleted functions with varying relative abundance and expression levels between healthy and HLB rhizoplane samples. Dark gray: rhizoplane-enriched function; light blue: rhizoplane-depleted; genes in green: increased relative abundance at the DNA level; genes in purple: increased expression level; genes in red: increased abundance and expression level. HLB.P (blue): rhizoplane samples from HLB trees; H.P (red): rhizoplane samples from healthy trees; HLB.S (green): rhizosphere samples from HLB trees; H.S (yellow): rhizosphere samples from healthy trees
QseC, an important quorum-sensing gene controlling bacterial flagella and motility [66], was rhizoplane-enriched and exhibited significantly increased abundance and expression levels in the healthy rhizoplane microbiome compared to the HLB samples. A similar pattern was observed for multiple root surface niche adaption-associated genes, such as bacterial chemotaxis sensor-related genes (including aer (aerotaxis (oxygen-sensing) receptor), tsr (serine receptor), tar (aspartate/maltose receptor), and trg (ribose and galactose chemoreceptor)); flagellar assembly-associated genes (Fig. 2c and Additional file 1: Figure S8); the surface contact signal sensing and biofilm formation regulation-associated genes wspABCDR [67]; and the chemosensory complex genes involved in pilus synthesis, including pilILJ, pilus assembly genes (Fig. 2c and Additional file 1: Figure S9), and LPS synthesis and transporter genes (Fig. 2c and Additional file 1: Figure S10). Multiple genes belonging to the secretion systems, including T1SS (hlyBD and raxAB), T2SS (gspCFHIJLM), T3SS (yscCJRSTUVNQ), T4aSS (virD4 and virB1-11 except virB7), and T4bSS (icmQ) and T6SS (ppkA, clpV, impKL, vasD and hcp, and its secreted substrate vgrG), were enriched on the rhizoplane. The majority of the identified secretion genes exhibited significantly increased relative abundance and expression activity in healthy rhizoplane samples compared to their HLB counterparts (Figs. 2c and 3). Concomitantly, effector genes, including xopAS, nolX, and hpa2, and several Rhs element vgr family genes exhibited significantly increased relative abundance and expression levels in the healthy rhizoplane microbiome compared to HLB samples (Fig. 2c).

The relative abundance and expression profiling of rhizoplane-enriched genes involved in bacterial secretion systems. Affiliation of each KO with secretion system types (I–VI) is indicated in brackets based on KEGG annotation. Red denotes “higher in healthy samples,” while blue denotes “higher in HLB samples.” The asterisk denotes P < 0.01; the plus sign denotes P < 0.05. MG, metagenome data, MT metatranscriptome data, P rhizoplane, S rhizosphere
The yesM-yesN two-component system, which is involved in carbohydrate utilization for bacteria [68], was enriched from the rhizosphere to rhizoplane microbiome. Several transporter-encoding genes whose products are responsible for importing plant-derived polysaccharide sources to bacterial cells including togBMNA (pectin-associated), fucP (fucose-associated), dgoT (d-galactonate-associated), araJ (arabinose-associated), nagE (N-acetylglucosamine-associated), lacE (lactose-associated), and dctS (C4-dicarboxylate sensor kinase) were enriched in the rhizoplane microbiome. Furthermore, the relative abundance of all these genes (expect togM) was increased in healthy rhizoplane samples. Significantly, increased expression was observed for togBNA and dctS in the healthy rhizoplane microbiome compared to HLB samples. Consistent with this observation, genes involved in “starch and sucrose metabolism” were enriched in the rhizoplane and exhibited increased abundance and expression levels in the healthy rhizoplane microbiome compared to HLB samples (Fig. 2c and Additional file 1: Figure S11). In contrast, genes involved in “carbon fixation pathways in prokaryotes,” which were depleted in the rhizoplane microbiome, exhibited increased relative abundance and expression activity in HLB rhizoplane samples (Figs. 2c and 4). Increased relative abundance was observed for nitrification-associated key genes (identified as rhizosphere- to rhizoplane-depleted), including amoA, amoB, amoC, and hao (hydroxylamine oxidoreductase), in HLB rhizoplane samples compared to healthy samples. The urease-encoding genes ureA, ureB, and ureC involved in degrading urea to ammonia, which contributes to both nitrification and carbon fixation, and the transporter genes for the coenzyme nickel metal for the urease [69], nikA and nikB, also exhibited significantly increased expression levels in HLB rhizoplane samples compared to healthy samples (Fig. 2c and Additional file 1: Figure S12).

Relative abundance and expression profiling of the rhizoplane-depleted genes involved in carbon fixation. The affiliation of each KO with different carbon fixation pathways is indicated in brackets based on KEGG annotation. CC Calvin cycle, RCAC reductive citric acid cycle, RAC reductive acetyl-CoA pathway, PCC/ACC 3-hydroxypropionate bicycle, HHC hydroxypropionate-hydroxybutyrate cycle. Red denotes “higher in healthy samples,” while blue denotes “higher in HLB samples.” The asterisk denotes P < 0.01; the plus sign denotes P < 0.05. MG metagenome data, MT metatranscriptome data, P rhizoplane, S rhizosphere
Linking the rhizoplane-enriched taxonomic and functional properties
To visualize the association between rhizoplane-enriched taxonomic and functional properties, we determined the taxonomic origin of rhizoplane-enriched functional attributes for all samples. The rhizoplane-enriched KOs were clustered at KEGG pathway level 3, and the clustered categories involved in host-microbe interactions identified in the above section, including the bacterial secretion system (BSS), flagellar assembly (FA), pilus assembly (PA), bacterial chemotaxis (BCh), and LPS synthesis (LPS), were analyzed, as were the rhizoplane-enriched KOs involved in starch and sucrose metabolism (SSM). Proteobacteria were the main contributor of these functions and contributed significantly more to the rhizoplane samples (95.3 ± 0.15% of the normalized total relative contribution for the six functional categories) than the rhizosphere samples (86.6 ± 1.27%) (paired t test, p < 0.01). A reduced contribution by taxa belonging to Acidobacteria and Actinobacteria was also observed (two taxa together accounting for 2.98 ± 0.14% of the normalized total relative contribution for the rhizoplane samples and 9.59 ± 1.43% for the rhizosphere samples, paired t test, p < 0.05) (Fig. 5). The relative contribution of Bradyrhizobium and Burkholderia to the six functional categories for the rhizoplane samples ranged from 6.2 to 38.1% and 2.9 to 19.2%, respectively, and their relative contribution to the rhizoplane samples significantly increased compared to the rhizosphere samples (p < 0.05 for Bradyrhizobium and p = 3.4E−5 for Burkholderia, respectively). The genes involved in the type IVb secretion system identified from our samples were mostly from unclassified bacteria (Additional file 1: Figure S13).

Relative contribution of different taxa (family or genus levels) to identified rhizoplane-enriched functional attributes in different samples. BSS bacterial secretion system, FA flagellar assembly, PA pilus assembly, BCh bacteria chemotaxis, LPS lipopolysaccharide assembly, SSM starch and sucrose metabolism
Expression activity comparison of Bradyrhizobium and Burkholderia between rhizosphere and rhizoplane samples
Bradyrhizobium and Burkholderia were the top two dominant and rhizoplane-enriched genera. To reveal the mechanisms underlying their highly efficient colonization, particularly in the rhizoplane, we extracted all genes originating from the two genera from the KO-annotated genes. In all, 2228 Bradyrhizobium and 2603 Burkholderia KOs were identified. These identified KOs exhibited overall higher relative abundance in the rhizoplane microbiome than the rhizosphere microbiome on a functional potential level (Additional file 1: Figure S14), further demonstrating rhizosphere-to-rhizoplane enrichment for both genera. The differentially expressed (DE) KOs between the rhizoplane and rhizosphere samples for the two genera were further determined using metatranscriptomic data. In total, 287 and 185 DE KOs were identified for Bradyrhizobium and Burkholderia, respectively, including 163 rhizoplane-upregulated and 124 rhizoplane-downregulated KOs for Bradyrhizobium and 89 rhizoplane-upregulated and 96 rhizoplane-downregulated KOs for Burkholderia. The majority of these rhizoplane-upregulated KOs (153 of 163 for Bradyrhizobium and 83 of 89 for Burkholderia) were not identified when comparing the rhizoplane and rhizosphere samples at the whole community level using metatranscriptomic data. Overrepresentation of multiple KEGG pathways involved in metabolism was observed for the rhizoplane-downregulated KOs for Bradyrhizobium and Burkholderia, and consistent enrichment of the categories “energy metabolism” and “carbohydrate metabolism” for both genera was also observed (p < 0.05, Fisher’s exact test, two-tail). The Bradyrhizobium rhizoplane-upregulated KOs were enriched in the categories of “transcription” and “metabolism|unclassified,” and the Burkholderia rhizoplane-upregulated KOs were enriched in the categories of “signal transduction” and “cell motility” (Table 1). All Burkholderia rhizoplane-upregulated KOs involved in “cell motility” were responsible for flagellar and pilus assembly; the KO representing the flagellar transcriptional activator flhD was found in the “signal transduction” category. Several other efflux system-associated genes, such as mdtA and mdtC, were also present in the Burkholderia “signal transduction” category. The majority of the Bradyrhizobium rhizoplane-upregulated KOs belonging to “metabolism|unclassified” were associated with cell wall synthesis. Interestingly, the gene encoding salicylate hydroxylase which converts salicylic acid, a critical phytohormone for plant systemic acquired resistance, to nonfunctional compound catechol was expressed at a significantly increased level in the rhizoplane compared to the rhizosphere for Bradyrhizobium.
Effects of inoculated Burkholderia strains on plant fitness
Members of Burkholderia have been known to benefit plants. Because Burkholderia are enriched from the rhizosphere to rhizoplane and it is one of the most abundant bacteria associated with citrus roots, we determined the contribution of Burkholderia to the citrus hosts. We isolated multiple Burkholderia strains from the rhizosphere of healthy citrus plants in a relevant study (Riera et al., unpublished data). We selected two representative strains A53 (Burkholderia metallica) and A63 (Burkholderia territori), which showed the best antagonistic activities against Sinorhizobium meliloti, a relative of the HLB causal agent Las, and several other citrus pathogens, such as Phytophthora spp. and Alternaria alternate (Riera et al., unpublished data), to inoculate citrus plants using the soil drench method. The results demonstrated that the two strains successfully colonized the root surface and maintained significantly higher relative abundance inside the rhizoplane bacterial community than inside the rhizosphere bacterial community (Student’s t test, p < 0.05 for both strains when detected at 5 and 9 dpi) (Additional file 1: Figure S15), demonstrating that the two strains were more adapted to the rhizoplane niche than that to the rhizosphere. The inoculated strains survived well, even 7 months after inoculation (Figs. 6a, b). We then conducted a greenhouse study to evaluate the effects of the selected strains on plant fitness. Salicylic acid (SA)-mediated induced systemic resistance (ISR) is an important benefit of beneficial bacteria to the plant host [70]. We determined the expression of three SA-mediated ISR marker genes, SAM, PR1, and PR2, for the inoculated trees. Plants treated with strain A53 exhibited a significant upregulation of PR2 gene at 3 dpi compared to negative control plants. A63 induced expression of the SAM gene at 5 dpi and the PR1 gene at 7 dpi. Similarly, Actigard, an analogue of SA, induced PR1 and SAM gene expression at 5 and 7 dpi (Fig. 6c).

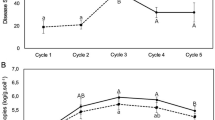
Survival rates of Burkholderia spp. inoculation and effects on expression of the citrus ISR-associated genes. Colony-forming units were represented in log10 scale for a strain A53 and b strain A63. Colonies were counted in NA plates supplemented with kanamycin (50 μg/mL) since both strains are kanamycin-resistant. Days post inoculation (dpi) was calculated up to 225, approximately 7 months post inoculation. Error bars represent standard deviation for three biological repeats performed independently. c Relative gene expression of three defense-related citrus genes PR1, PR2, and SAM. Fold change of gene expression is represented at 3, 5, and 7 days post inoculation in root system. Fold change was calculated using ∆∆Ct method. Boxplots represent fold change compared to non-treated plants for three biological replicates. Each biological replicate contains three technical repeats
Discussion
Our data demonstrated reduced genetic and microbial complexity in the citrus rhizoplane microbiome compared to that in the rhizosphere communities, indicating the filter effect of plant hosts on the closely associated rhizoplane microbiome assembly [5]. This finding is consistent with previous findings based on annual plants [13, 17], indicating that the rhizosphere-to-rhizoplane enrichment of microbiome occurs in both annual and perennial plants. Our data further suggested that the niche enrichment process is fine-tuned by the plant host for the desired functions, resulting in dramatic structural changes of the microbiome (Fig. 5). Bacteria were the most dominant domain and accounted for more than 99% of the citrus root-associated microbiome, whereas minimal Archaea and fungi were found. The taxonomic composition of the citrus root-associated microbiome identified here was very similar with that of wheat and cucumber, where the relative abundance of bacteria is more than 99% [15]. Proteobacteria, which was dominated by Bradyrhizobium and Burkholderia (Fig. 1), was enriched on the rhizoplane, and depletion of Actinobacteria and Acidobacteria was observed in this process. We demonstrated that the genes involved in utilizing the root-derived carbon source were enriched from the rhizosphere to the rhizoplane, further suggesting the importance of plant cell wall polysaccharide utilization-associated genes for the rhizoplane microbiome. These genes were gradually enriched from the free living bulk soil inoculum, the rhizosphere to the rhizoplane [15]. However, the genes involved in “carbon fixation pathways in prokaryotes,” which are critical for autotrophic microorganisms, were depleted in rhizoplane samples, collectively highlighting that the plant-derived carbon sources are an important driving force for citrus rhizoplane microbiome assembly. This finding is consistent with the notion that the microbiome on the rhizoplane enjoys superior access to plant exudates compared to the microbiome in the rhizosphere.
Functional features, such as motility, chemotaxis, two-component system and secretion systems, LPS, and type IV pilus synthesis, were rhizoplane-enriched, indicating more active plant-microbe interactions on the rhizoplane than on the rhizosphere. We envision that following chemotaxis sensing, the active motility mediated mainly by flagella and pili allows the microbes to rapidly reach the preferred root surface niches to form biofilms or aggregates with the help of adhesions such as LPS and pili. Interestingly, several secreted effectors were actively expressed in the rhizoplane microbiome that may benefit the microbiome by modulating the plant immune system. Rhizoplane enrichment of Bradyrhizobium and Burkholderia was observed in this study. Similar enrichment patterns were also observed when analyzing samples collected from 27 important citrus-growing areas worldwide, suggesting the ubiquity and importance of these two genera for the citrus root-associated microbiome (Zhang and Wang, unpublished data). The enrichment of Bradyrhizobium and Burkholderia on the rhizoplane indicated their successful strategy for rhizoplane adaption. The metatranscriptomic data demonstrated that the functions involved in metabolic activities were more actively expressed in the rhizosphere than the rhizoplane for both genera, further suggesting that nutrient resources were more easily accessed for the rhizoplane microbiome than the rhizosphere microbiome. In addition to common rhizoplane-enriched genes, multiple Bradyrhizobium and Burkholderia specific genes, such as efflux system-associated genes, cell motility-associated genes, and plant defense-resistant-associated genes, were more active on the rhizoplane than the rhizosphere for Bradyrhizobium and Burkholderia but were not identified for the remaining microorganisms. The efflux system-associated genes contribute to successful interactions with host plants [71]. The salicylic acid-degrading enzyme salicylate hydroxylase encoding gene from Bradyrhizobium exhibited significantly increased expression activity on the rhizoplane and might contribute to the colonization of Bradyrhizobium by suppressing the plant defense response [72].
The relative abundance of the rhizoplane-enriched taxa and functional properties, as well as the expression profiling of the rhizoplane-enriched functional features were significantly reduced by HLB. Bradyrhizobium and Burkholderia, the two most dominant rhizoplane-enriched genera, exhibited reduced relative abundance in HLB rhizoplane samples compared to healthy samples. Our previous cultivation-based results demonstrated that the Burkholderia population was significantly increased in the healthy citrus endorhiza microbiome compared to HLB samples, whereas Bradyrhizobium was not observed, mainly because of their slow growth (~2 weeks for colony formation. This is longer than the experiment design, during which colonies were harvested after 3–10 days of incubation) [22]. These results suggested that the citrus plants prefer Bradyrhizobium and Burkholderia and enrich them from the rhizosphere to rhizoplane. The survival rate assay of the two inoculated Burkholderia strains further demonstrated that the Burkholderia strains were more adapted to the rhizoplane niche than the rhizosphere. Root-associated Bradyrhizobium is frequently reported as plant-beneficial bacteria [55, 73]; beneficial effects of Burkholderia also are frequently observed [22, 56, 74]. Furthermore, when the citrus were inoculated with Burkholderia strains isolated from the healthy citrus root-associated microbiome, several ISR-associated genes in the citrus trees were induced (Fig. 6) that might reduce HLB disease progress. Multiple beneficial bacteria stimulate plant immunity in an SA-dependent ISR manner [70]. The observed upregulation of ISR-associated genes, especially the upregulation of SAM by Burkholderia sp. inoculation, likely promotes plant health by triggering SA-dependent broad-spectrum systemic resistance to pathogens, which might be an important aspect of plant-beneficial microbe interactions. Other rhizoplane-enriched taxa, such as Variovorax and Bdellovibrio, exhibited decreased relative abundance in the rhizoplane microbiome when trees became infected with Las (Fig. 1). Variovorax and Bdellovibrio exhibit plant growth promotion effects [57, 58]. Additionally, our results revealed rhizoplane-enriched functional properties, including chemotaxis, flagellar assembly, LPS synthesis and transport, as well as secretion system and related effectors, were depleted by HLB. However, the rhizoplane-depleted functional features, including “carbon fixation pathways in prokaryotes” and nitrification and denitrification-related genes, exhibited significantly increased relative abundance and expression levels in the HLB rhizoplane microbiome compared to those in the healthy samples (Figs. 2, 3, and 4 and Additional file 1: Figures S8–S13). Reduced relative abundance and activity of plant-derived polysaccharide utilization-associated genes together with the increased relative abundance and activity of autotrophic life cycle-adapted carbon fixation, nitrification, and denitrification functional features, support our notion that HLB trees supplied less easy-to-use carbon source, e.g., sucrose for root-associated microbiome use [24]. These data are consistent with the notion that HLB affects the availability of sucrose to microbes on the rhizoplane because of its effect on phloem transportation of photoassimilates [18] and root decline [20]. In particular, HLB affects functional features, such as motility, chemotaxis, two-component system and secretion systems, as well as LPS and type IV pilus synthesis with significantly increased relative abundance and expression levels in the healthy rhizoplane microbiome compared in HLB samples (Fig. 2). These findings clearly demonstrated the negative effect on the microbiome at the community level. The negative effects of HLB on the citrus microbiome will ultimately deteriorate the beneficial interactions between the microbiome and the host.
Conclusions
Overall, we demonstrate that the functional properties involved in host-microbe interactions are critical for the microbiome-inhabiting plant root surfaces and are influenced dramatically by the availability of plant exudates. These rhizoplane-enriched functional properties can subsequently benefit the plant host. HLB not only alters the physiology of the citrus host but also impairs the microbiome-host interaction. Our study provides novel insights for understanding the composition and function of the plant rhizoplane-enriched microbiome and its effect on plant health.
Abbreviations
- AGS:
-
Average genome size
- CFU:
-
Colony-forming unit
- dpi:
-
Days post inoculation
- HLB:
-
Huanglongbing
- ISR:
-
Induced systemic resistance
- KO:
-
KEGG orthologue
- MG:
-
Metagenome
- MT:
-
Metatranscriptome
- SA:
-
Salicylic acid
- VPA:
-
Variation partitioning analysis
References
Turner TR, James EK, Poole PS. The plant microbiome. Genome Biol. 2013;14:209.
Schlaeppi K, Bulgarelli D. The plant microbiome at work. Mol Plant-Microbe Interact. 2015;25:212–7.
Wang N, Jin T, Trivedi P, Setubal JC, Tang J, A. MM, et al. Announcement of the international citrus microbiome (Phytobiome) consortium. J Citrus Pathol. 2015:2.
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A. The importance of the microbiome of the plant holobiont. New Phytol. 2015;206:1196–206.
Reinhold-Hurek B, Bünger W, Burbano CS, Sabale M, Hurek T. Roots shaping their microbiome: global hotspots for microbial activity. Annu Rev Phytopathol. 2015;53:403–24.
Lebeis SL. Greater than the sum of their parts: characterizing plant microbiomes at the community-level. Curr Opin Plant Biol. 2015;24:82–6.
Panke-Buisse K, Poole AC, Goodrich JK, Ley RE, Kao-Kniffin J. Selection on soil microbiomes reveals reproducible impacts on plant function. ISME J. 2015;9:980–9.
Karasov TL, Kniskern JM, Gao L, DeYoung BJ, Ding J, Dubiella U, et al. The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature. 2014;512:436–40.
Sessitsch A, Mitter B. 21st century agriculture: integration of plant microbiomes for improved crop production and food security. Microb Biotechnol. 2015;8:32–3.
van der Heijden MGA, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, et al. Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature. 1998;396:69–72.
Torsvik V. Prokaryotic diversity—magnitude, dynamics, and controlling factors. Science. 2002;296:1064–6.
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488:86–90.
Bulgarelli D, Rott M, Schlaeppi K, Ver Loren van Themaat E, Ahmadinejad N, Assenza F, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 2012;488:91–5.
Mendes LW, Kuramae EE, Navarrete AA, van Veen JA, Tsai SM. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014;8:1577–87.
Ofek-Lalzar M, Sela N, Goldman-Voronov M, Green SJ, Hadar Y, Minz D. Niche and host-associated functional signatures of the root surface microbiome. Nat Commun. 2014;5:4950.
Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, et al. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015;17:392–403.
Edwards J, Johnson C, Santos-Medellín C, Lurie E, Podishetty NK, Bhatnagar S, et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc Natl Acad Sci. 2015;112:E911–20.
Wang N, Trivedi P. Citrus Huanglongbing: a newly relevant disease presents unprecedented challenges. Phytopathology. 2013;103:652–65.
Gottwald TR. Current epidemiological understanding on citrus Huanglongbing. Annu Rev Phytopathol. 2010;48:119–39.
Johnson EG, Wu J, Bright DB, Graham JH. Association of “Candidatus Liberibacter asiaticus” root infection, but not phloem plugging with root loss on huanglongbing-affected trees prior to appearance of foliar symptoms. Plant Pathol. 2014;63:290–8.
Sagaram US, Deangelis KM, Trivedi P, Andersen GL, Lu SE, Wang N. Bacterial diversity analysis of huanglongbing pathogen-infected citrus, using phyloChip arrays and 16S rRNA gene clone library sequencing. Appl Environ Microbiol. 2009;75:1566–74.
Trivedi P, Spann T, Wang N. Isolation and characterization of beneficial bacteria associated with citrus roots in Florida. Microb Ecol. 2011;62:324–36.
Trivedi P, Duan YP, Wang NA. Huanglongbing, a systemic disease, restructures the bacterial community associated with citrus roots. Appl Environ Microbiol. 2010;76:3427–36.
Trivedi P, He ZL, Van Nostrand JD, Albrigo G, Zhou JZ, Wang N. Huanglongbing alters the structure and functional diversity of microbial communities associated with citrus rhizosphere. ISME J. 2012;6:363–83.
Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. 2011; Available at https://github.com/najoshi/sickle.
Zhang Y, Barthe GA, Grosser J, Wang N. Transcriptome analysis of root response to citrus blight based on the newly assembled Swingle citrumelo draft genome. BMC Genomics. 2016;17(1):485.
Xu Q, Chen L-L, Ruan X, Chen D, Zhu A, Chen C, et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet. 2013;45:59–66.
Wu GA, Prochnik S, Jenkins J, Salse J, Hellsten U, Murat F, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat Biotechnol. 2014;32:656–62.
Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–9.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38:e132.
Fu LM, Niu BF, Zhu ZW, Wu ST, Li WZ. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res. 2007;17:377–86.
Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14:927–30.
Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35.
Warnes GR, Bolker B, Bonebakker L, Gentleman R, Liaw WHA, Lumley T, et al. Gplots: various R programming tools for plotting data. R Package. version 2.17.0. 2015; Available from: http://cran.r-project.org/package=gplots
Nayfach S, Pollard KS. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 2015;16:51.
Bart R, Cohn M, Kassen A, McCallum EJ, Shybut M, Petriello A, et al. High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc Natl Acad Sci. 2012;109:E1972–9.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer J. 2015;3:e1165.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014;42(D1): D206-D214.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid tech. Bact. Syst. Wiley: Chichester; 1991. p. 125–75.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Fierer N, Jackson JA, Vilgalys R, Jackson RB. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol. 2005;71:4117–20.
Francis MI, Redondo A, Burns JK, Graham JH. Soil application of imidacloprid and related SAR-inducing compounds produces effective and persistent control of citrus canker. Eur J Plant Pathol. 2009;124:283–92.
Kopylova E, Noé L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7.
Uchiyama I, Mihara M, Nishide H, Chiba H. MBGD update 2015: microbial genome database for flexible ortholog analysis utilizing a diverse set of genomic data. Nucleic Acids Res. 2015;43:D270–6.
Coenye T, Goris J, Spilker T, Vandamme P, LiPuma JJ. Characterization of unusual bacteria isolated from respiratory secretions of cystic fibrosis patients and description of Inquilinus limosus gen. Nov., sp. nov. J Clin Microbiol. 2002;40:2062–9.
Schröttner P, Rudolph WW, Taube F, Gunzer F. First report on the isolation of Aureimonas altamirensis from a patient with peritonitis. Int J Infect Dis. 2014;29:e71–3.
Rouws LFM, Leite J, de Matos GF, Zilli JE, Coelho MRR, Xavier GR, et al. Endophytic Bradyrhizobium spp. isolates from sugarcane obtained through different culture strategies. Environ Microbiol Rep. 2014;6:354–63.
Bernabeu PR, Pistorio M, Torres-Tejerizo G, Estrada-De los Santos P, Galar ML, Boiardi JL, et al. Colonization and plant growth-promotion of tomato by Burkholderia tropica. Sci Hortic (Amsterdam). 2015;191:113–20.
Satola B, Wübbeler JH, Steinbüchel A. Metabolic characteristics of the species Variovorax paradoxus. Appl Microbiol Biotechnol. 2013;97:541–60.
Marupakula S, Mahmood S, Finlay RD. Analysis of single root tip microbiomes suggests that distinctive bacterial communities are selected by Pinus sylvestris roots colonized by different ectomycorrhizal fungi. Environ Microbiol. 2016;18(5):1470–83.
Raupach GS, Kloepper JW. Mixtures of plant growth-promoting rhizobacteria enhance biological control of multiple cucumber pathogens. Phytopathology. 1998;88:1158–64.
Xu X-M, Jeffries P, Pautasso M, Jeger MJ. Combined use of biocontrol agents to manage plant diseases in theory and practice. Phytopathology. 2011;101:1024–31.
Haichar FEZ, Marol C, Berge O, Rangel-Castro JI, Prosser JI, Balesdent J, et al. Plant host habitat and root exudates shape soil bacterial community structure. ISME J. 2008;2:1221–30.
Wakelin SA, Warren RA, Harvey PR, Ryder MH. Phosphate solubilization by Penicillium spp. closely associated with wheat roots. Biol Fertil Soils. 2004;40:36–43.
Anderson M, Habiger J. Characterization and identification of productivity-associated rhizobacteria in wheat. Appl Environ Microbiol. 2012;78:4434–46.
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. DbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(W1):W445-451.
Zhang L-M, Offre PR, He J-Z, Verhamme DT, Nicol GW, Prosser JI. Autotrophic ammonia oxidation by soil thaumarchaea. Proc Natl Acad Sci U S A. 2010;107:17240–5.
Sperandio V, Torres AG, Kaper JB. Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol Microbiol. 2002;43:809–21.
Spiers AJ, Kahn SG, Bohannon J, Travisano M, Rainey PB. Adaptive divergence in experimental populations of Pseudomonas fluorescens. I. Genetic and phenotypic bases of wrinkly spreader fitness. Genetics. 2002;161:33–46.
Xu W, Zhang Y, Huang M, Yi X, Gao X, Zhang D, et al. The yesN gene encodes a carbohydrate utilization regulatory protein in lactobacillus plantarum. Ann Microbiol. 2014;65:115–20.
Lv J, Jiang Y, Yu Q, Lu S. Structural and functional role of nickel ions in urease by molecular dynamics simulation. J Biol Inorg Chem. 2011;16:125–35.
Pieterse CMJ, Zamioudis C, Berendsen RL, Weller DM, Van Wees SCM, Bakker PAHM. Induced systemic resistance by beneficial microbes. Annu Rev Phytopathol. 2014;52:347–75.
Martinez JL, Sánchez MB, Martínez-Solano L, Hernandez A, Garmendia L, Fajardo A, et al. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol Rev. 2009; 33:430–49.
Delaney TP, Uknes S, Vernooij B, Friedrich L, Weymann K, Negrotto D, et al. A central role of salicylic acid in plant disease resistance. Science. 1994;266:1247–50.
Thaweenut N, Hachisuka Y, Ando S, Yanagisawa S, Yoneyama T. Two seasons’ study on nifH gene expression and nitrogen fixation by diazotrophic endophytes in sugarcane (Saccharum spp. hybrids): expression of nifH genes similar to those of rhizobia. Plant Soil. 2011;338:435–49.
Suárez-Moreno ZR, Caballero-Mellado J, Coutinho BG, Mendonça-Previato L, James EK, Venturi V. Common features of environmental and potentially beneficial plant-associated Burkholderia. Microb Ecol. 2012;93:249–66.
Acknowledgements
We would like to thank Utpal Handique for the help in selecting healthy and HLB trees.
Funding
This study has been supported by Florida Citrus Research and Development Foundation.
Availability of data and materials
The DNA and RNA reads have been deposited at NCBI under the bioproject accession no. PRJNA324090 and SRA accession no. SRP076109.
Author information
Authors and Affiliations
Contributions
NW conceived and supervised the project. YZ and NW designed the experiment. YZ and NR collected the samples and extracted nucleic acid. YZ and JX analyzed the data. NR conducted the bacterial inoculation and plant gene expression experiments. NW, YZ JX, NR, and TJ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
No permissions and/or licenses are required since citrus is not an endangered species.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Summary of the metagenome data. Table S2. Summary of the metagenome assembly and unigenes. Table S3. Summary of DNA and RNA reads mapping results. Table S4. The alpha diversity (Shannon index) of the microbial communities. Table S5. The relationship between the MAG Cellvibrio bin.79 and related genomes. Table S6. The relationship between the MAG Thaumarchaeota bin.105 and related genomes. Figure S1. The visual symptoms and titer of ‘Ca. L. asiaticus’ in root and leaf samples of HLB symptomatic and healthy citrus trees. Figure S2. Taxonomic composition of citrus root-associated microbiome. Figure S3. The taxonomic composition of citrus root-associated microbiome. Figure S4. The rhizoplane-enriched genera with different relative abundance between healthy and HLB rhizoplane samples. Figure S5. (A) The relative abundance of Cellvibrio among all the samples. (B) The expression activity of IAA producing related genes in bin.74. Figure S6. The rhizosphere- to rhizoplane-depleted genera with different relative abundance between healthy and HLB rhizoplane samples. Figure S7. (A) The relative abundance of Thaumarchaeota among all the samples. (B) The phylogenetic position of the bin.105 based on amoA gene. Figure S8-S12. The relative abundance and expression profiling of genes involved in flagellar assembly, pilus assembly, LPS assembly, starch and sucrose, and nitrogen metabolism, respectively. Figure S13. Relative contribution of different taxa to the identified rhizoplane-enriched genes involved in type IVb secretion system. Figure S14. The relative abundance of Bradyrhizobium (A) and Burkholderia (B) KOs among all samples. Figure S15. The population dynamic of the inoculated strains in the rhizosphere and rhizoplane bacterial community of the inoculated citrus plants. (DOCX 18086 kb).
Additional file 2:
The expression activity of the identified rhizoplane-enriched KOs. Pr denotes the sample is from rhizoplane samples, while Sr denotes the sample is from rhizosphere samples. Samples 1–3 are from healthy trees, and samples 4–6 are from HLB trees. The reads count is shown in the table. (XLSX 233 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., Xu, J., Riera, N. et al. Huanglongbing impairs the rhizosphere-to-rhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 5, 97 (2017). https://doi.org/10.1186/s40168-017-0304-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-017-0304-4