
Abstract
Background
In recent years, research in artificial neural networks has resurged, now under the deep-learning umbrella, and grown extremely popular. Recently reported success of DL techniques in crowd-sourced QSAR and predictive toxicology competitions has showcased these methods as powerful tools in drug-discovery and toxicology research. The aim of this work was dual, first large number of hyper-parameter configurations were explored to investigate how they affect the performance of DNNs and could act as starting points when tuning DNNs and second their performance was compared to popular methods widely employed in the field of cheminformatics namely Naïve Bayes, k-nearest neighbor, random forest and support vector machines. Moreover, robustness of machine learning methods to different levels of artificially introduced noise was assessed. The open-source Caffe deep-learning framework and modern NVidia GPU units were utilized to carry out this study, allowing large number of DNN configurations to be explored.
Results
We show that feed-forward deep neural networks are capable of achieving strong classification performance and outperform shallow methods across diverse activity classes when optimized. Hyper-parameters that were found to play critical role are the activation function, dropout regularization, number hidden layers and number of neurons. When compared to the rest methods, tuned DNNs were found to statistically outperform, with p value <0.01 based on Wilcoxon statistical test. DNN achieved on average MCC units of 0.149 higher than NB, 0.092 than kNN, 0.052 than SVM with linear kernel, 0.021 than RF and finally 0.009 higher than SVM with radial basis function kernel. When exploring robustness to noise, non-linear methods were found to perform well when dealing with low levels of noise, lower than or equal to 20%, however when dealing with higher levels of noise, higher than 30%, the Naïve Bayes method was found to perform well and even outperform at the highest level of noise 50% more sophisticated methods across several datasets.
Similar content being viewed by others

Background
Machine learning techniques have become an integral part of the modern drug discovery process. Data mining methods based on machine learning techniques are routinely applied to model complex physicochemical and chemo-biological endpoints. Applications range from quantitative structure–property relationships (QSPRs) [1,2,3], quantitative structure–activity relationships (QSARs) [4, 5] to in silico mode-of-action analysis and predictive toxicology [6, 7].
Artificial neural networks (ANNs) were once popular in the field of molecular informatics and have been applied for wide range of QSARs/QSPRs applications [8,9,10,11,12,13,14,15,16,17]. With the introduction of shallow methods such as random forest and support vector machines and technical difficulties associated with ANNs led to gradual decrease of popularity of these methods. In recent years, research in ANNs has resurged, now under the deep-learning umbrella, and grown extremely popular due to major breakthroughs in computing capabilities, attracting considerable attention from both academia and industry. Although reported applications of deep-learning techniques still remain limited in the field of molecular modeling they are fast gaining attention [18]. Two major crowd-sourced competitions, the Merck QSAR competition, held in 2012 at the Kaggle data mining platform, and the Tox21 data-challenge 2014 for chemical risk assessment were both won by groups that utilized deep learning methods as main machine learning techniques [19, 20]. The recent success demonstrates that deep learning methods are well suited for modeling complex biological data to support drug discovery and toxicological research.
ANNs were designed to mimic the efficiency and robustness of biological systems inspired by the complex cerebral cortex to process the myriads amount of sensory data [21]. ANNs attempt to represent highly non-linear and varying functions by combining multiple levels of representations through interconnected non-linear transformations [22]. Deep-learning methods are part of distributed representation-learning algorithms that attempt to extract and organize discriminative information from the data by discovering features that compose multi-level distributed representations [23]. Representation learning algorithms have been applied with success to model complex real-world problems, such as speech recognition, signal processing and image classification [21, 24,25,26].
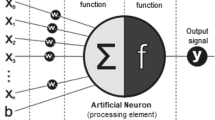
An DNN consists of multiple fully-connected layers: an input layer, one or multiple hidden layers and a single output layer [27]. In feed-forward DNN, one of the mainstream deep learning methods, information is flowing forward from the input layer, through the hidden layers and towards the output layer, illustration shown in Fig. 1a. The building block of DNN is the artificial neuron, which was introduced in 1943 by McCulloh and Pitts [28] and was inspired by biological neurons, illustration shown in Fig. 1b. Each neuron receives one or more input signals x 1, x 2, …, x m and outputs a value y to neurons of the next layer and so forth. The output y is a nonlinear weighted sum of input signals, Fig. 1c shows popular activation functions the rectified linear units (ReLU), Tanh and Sigmoid (Sigm).

a A feed-forward deep neural network with two hidden layers, each layer consists of multiple neurons, which are fully connected with neurons of the previous and following layers. b Each artificial neuron receives one or more input signals x 1, x 2,…, x m and outputs a value y to neurons of the next layer. The output y is a nonlinear weighted sum of input signals. Nonlinearity is achieved by passing the linear sum through non-linear functions known as activation functions. c Popular neurons activation functions: the rectified linear unit (ReLU) (red), Sigmoid (Sigm) (green) and Tanh (blue)
Deep neural networks are typically trained, by updating and adjusting neurons weights and biases, utilizing the supervised learning back-propagation algorithm in conjunction with optimization technique such as stochastic gradient descent [29, 30]. Regularization techniques, such as Dropout, were introduced to reduce over-fitting and improve generalization [31]. Dropout regularization technique was proposed to address the problem of over-fitting by preventing neurons co-adaption. Recent advances in GPU computing capabilities has allowed complex deep learning architecture to be trained in reasonable amount of time [32].
Lusci et al. [33] were among first to investigate the application of deep neural architectures for modeling molecular properties. The authors utilized a recursive deep neural network approach to model molecular properties and applied the proposed approach to develop predictive model for aqueous solubility. Xu et al. [34] applied deep learning to model drug-induced liver injury. The authors developed a predictive model based on 475 drugs and applied on external dataset of 198 drugs achieving AUC of 0.955 and exceeding the performance of previously reported drug-induced liver injury models. Aliper et al. [35] applied deep learning for predicting pharmacological properties of drugs and for drug repurposing utilizing transcriptomic data from the LINCS Project. The authors demonstrated that DNNs trained on transcriptional response data sets could be utilized to classify drugs indications based on their transcriptional profiles. More recently, attempts to utilize convolutional neural networks (CNN) to model chemical data were also reported. Duvenaud et al., reported a CNN approach that operates directly on molecular graphs, where molecular structures are represented as graphs of arbitrary size and shape [36]. CNN were reported to be able to learn neural graph fingerprints and even outperform standard circular fingerprints on several tested datasets.
Recent study by Ma et al. [19] investigated and compared the performance of deep neural networks to random forest for QSARs applications. The authors employed arbitrary selected DNNs configurations as opposed to an exhaustive search of possible hyper-parameters combinations, and compared the performance of DNNs against Random Forest for regression using 15 Merk’s in-house datasets including on-target and absorption, distribution, metabolism, and excretion (ADME) endpoints relevant to pharmaceutical research. The authors reported that over 50 DNN configurations were explored and the performance of DNN was compared to RF. DNN was reported to outperform RF overall by achieving on average 0.051 better R 2 across the 15 activity classes employed.
While several studies utilizing deep learning techniques have been recently reported in the literature, the use of customized in-house implementations, often based on Theano framework, and models often remain non accessible to readers, hence hindering wider adoption of deep learning techniques [37]. Instead, in this work we opted for the open-source Caffe deep learning framework as the main framework for training and testing deep neural networks configurations. Caffe provides an easy usable platform for defining DNN configurations and executing experiments without extensive customizable hard-coding required as it provides an expressive and highly modular architecture [38]. The code used to carry out this study, which was built upon Caffe and termed “ChemoCaffe”, is also made publicly available in the hope to make deep learning more accessible to practitioners interested applying DNNs to chemo-biological problems. ChemoCaffe allows large number of arbitrary selected DNN configurations to be tested in an automated fashion until optimal set of hyper-parameter values is identified.
Moreover, there is lack of comprehensive comparison of deep learning methods to popular methods of commonly employed algorithms such as random forest, support vector machine, k-nearest neighbor and Naïve Bayes for modeling structure–activity relationships (SARs). In this study we address this gap in the literature by conducting and reporting a comprehensive comparison utilizing seven diverse bioactivity classes extracted from ChEMBL public repository. Furthermore, it is commonly assumed that there is a limited amount of noise present in bioactivity datasets. There is no empirical evaluation of deep learning methods when dealing with noisy data, a common situation prevalent in Cheminformatics applications, especially when dealing with high-throughput screening readings. Hence, we explored the robustness of machine learning algorithms included in this study by introducing different levels of artificial noise to the datasets and measuring the performance of each method (Table 1).
Results and discussion
The experimental process followed consisted of two main parts. First, arbitrary but reasonable selected hyper-parameter configurations were explored with the aim to investigate how they affect the performance of feed-forward deep neural networks. In the second part of the study, comparison of performance of deep neural nets to shallow methods Bernoulli Naïve Bayes, k-nearest neighbor, random forest and support vector machine was investigated.
Optimizing deep neural networks hyper-parameters
Here the study was focused on the hyper-parameters: (a) activation functions, by comparing the performance of rectified linear unit (ReLU), Sigmoid (Sigm) and Tanh functions, (b) learning rate, (c) number of neurons per layer, (d) number of hidden layers and (e) dropout regularization. All tested network configurations were trained for 300 epochs with fixed learning rates, as shown in Table 2, with no early stops applied.
Comparing activation functions
Single hidden layered neural networks with varying number of neurons {5, 10, 50, 100, 200, 500, 700, 1000, 1500, 2000, 2500, 3000, 3500} were tested to compare the performance between the three activation functions ReLU, Sigm and Tanh, the rest hyper-parameters were kept fixed. The performance was measured using MCC over fivefolds cross validation. The results obtained per activity class and per activation function are presented in Fig. 2 with ReLU(red), Sigmoid (green) and Tanh (blue), obtained with learning rate of η = 0.1. Neural Networks combined with the ReLU activation function perform better, when compared to Sigm or Tanh, by achieving higher MCC. Statistical analysis using Wilcoxon paired rank test are shown in Table 3, with confidence interval 99%. The results demonstrate that DNN combined with the rectified linear units (ReLU) activation function performed better than the Sigm or Tanh functions. Neurons with rectified linear units (ReLU) activation function has been reported to be (a) easier to optimize, (b) converge faster, (c) generalize better and (d) are faster to compute [39].

Comparison of activation functions rectified linear units (ReLU), Tanh and Sigmoid (Sigm) on the performance of DNNs. DNNs with a single hidden layer and variable number of neurons were trained and tested using the activation functions ReLU (red), Sigm (green) and Tanh (blue) over fivefold cross validation. The performance was measured using MCC as evaluation metric
Number of hidden layers, neurons per layer and regularization
Feed-forward neural networks are considered “deep” when the number of hidden layers is equal to or more than two, where each layer represents a higher level of abstraction. Here the analysis was focused on the hyper-parameters; (a) number of hidden layers, (b) learning rate, (c) number of neurons per hidden layer and (d) “dropout” regularization, while retaining the rest hyper-parameters fixed. Experimental process followed was similar as before using internal fivefold cross validation.
Results obtained by seven deep neural nets configurations over the seven bioactivity classes are shown in Fig. 3, here the effect of hyper-parameters (a) number of hidden layers, (b) number of neurons and (c) dropout regularization on the performance of DNN measured by MCC as evaluation metric are visualized averaged over the seven activity classes, while the rest parameters were kept fixed. From these results it can be observed that increase of the number of hidden layers and neurons, combined with application of regularization, has a substantial effect on the performance of DNN. When comparing the improvement in performance observed between single hidden layered Neural Networks, configuration A, and deeper network configurations tested (B–G) with multiple hidden layers, thousands of neurons and regularization the performance was improved by an average of 0.062 MCC units. Furthermore, application of dropout regularization on the performance, configurations C–G, was also found to affect the performance, when comparing to non-regularized networks. As an example, the performance was improved by an average of 0.039 MCC units when dropout of 50% was applied to two hidden layered DNN. This highlights the importance of regularization on the performance of DNN.

Effect of the hyper-parameters (i) number of hidden layers, (ii) number of neurons and (iii) dropout regularization on the performance of DNNs measured by MCC as evaluation metric. DNN configuration A shows results obtained by DNN with a single hidden layer and 10 neurons, ReLU activation function and no regularization averaged over the seven activity datasets, B a two hidden layered DNN with 500 neurons in each layer, ReLU activation function and no regularization, C two hidden layers with 3000 neurons per hidden layer and dropout regularization (0% for the input and 50% for hidden layers), D two hidden layers with 3000 neurons per hidden layer and dropout regularization (20% for the input and 50% for hidden layers), E two hidden layers with 3000 neurons per hidden layer and dropout regularization (50% for both the input and hidden layers), F three hidden layers with 3000 neurons per layer and dropout regularization (50% for both the input and hidden layers) and G four hidden layers with 3500 neurons per layer and dropout regularization (50% for the input and hidden layers)
Comparison of performance of deep neural nets to shallow methods and robustness to noise
In the second part of the study comparison of performance of deep neural nets with the shallow counterparts Bernoulli Naïve Bayes (NB), k-nearest neighbor (kNN), random forest (RF) and support vector machines (SVM) was performed. Here the NB classifier was used as a baseline method. Each dataset was split to 60% for hyper-parameters tuning using fivefold cross validation and the rest 40% was used as validation set, with the experiments repeated three times for each dataset. Performance achieved by each algorithm on validation sets is presented in Table 4. Wilcoxon paired signed-rank statistical test was applied, with confidence intervals 99%, to compare the performance achieved by DNN against each of the rest methods, results are shown in Table 5.
The differences measured in MCC units between DNN and the rest algorithms are visualized in boxplot Fig. 4. Here DNN achieved on average MCC units of 0.149 higher than NB, 0.092 than kNN, 0.052 than SVM with linear kernel, 0.021 than RF and finally 0.009 higher than SVM with radial basis function kernel. These results demonstrate that deep neural nets, when tuned accordingly, are capable of achieving strong performance for modeling bioactivity data for small molecules and outperform popular machine learning methods when combined with simple circular fingerprints as molecular descriptors.

Boxplot of differences between performances achieved by tuned DNN and the rest algorithms measured using MCC as evaluation metric on the validation sets over the seven activity classes. Results are ranked by decreased mean differences. The differences ranged on average from 0.149 MCC units between DNN and NB, 0.092 DNN and kNN, 0.052 DNN and SVM with linear kernel, 0.021 DNN and RF and 0.009 DNN and SVM with “rbf” kernel
Structure–activity relationships collected from medicinal literature and stored in bioactivity databases may contain noise or erroneous reported activity measurements, due to human error or experimental uncertainty, that could potentially affect the performance of data-mining techniques [40,41,42,43]. Furthermore, results from high-throughput screenings (HTS) are known to suffer from false positive hits. Hence, we explored robustness of machine learning algorithms to noise by introducing artificial noise to the datasets and measuring the effect on the performance of the data mining techniques. Noise was introduced by randomly sampling and flipping the labels of a percentage of active instances onto inactive and vice versa equal number of randomly sampled inactive instances to active, in total five levels of noise were introduced in each activity dataset, ranging from 10 and up to 50%. This process was applied on the training sets, while the validation sets were not altered to serve as ground truth when measuring performance. The results from the process for 4 of the dataset are presented in Fig. 5. When examining robustness of machine learning algorithms to different levels of noise it was observed that non-linear methods overall outperformed Naïve Bayes when dealing with low levels of noise, lower or equal to 20%, where performance was retained equal to or higher than MCC of 0.7 on average. Instead, at higher level of noise equal or higher than 30% Naïve Bayes reached or even outperformed the rest methods, e.g. in cases of datasets CDK2 and CB1, demonstrating higher tolerance to noise. Naïve Bayes has been previously reported to perform well when dealing with noisy high-throughput screening data and achieve good enrichments among top retrieved compounds [44]. DNN overall outperformed the rest methods across most tested datasets, when dealt with noisy data their performance in several datasets dropped below that of other methods included in the study indicating that these methods might be more sensitive to noise that the rest methods. Hence when dealing with experimental datasets where low level of noise is expected non-linear methods are likely be a better option, e.g. measurements generated from confirmatory screening campaigns, instead when dealing with data where high level of noise is expected, e.g. high-throughput screening campaigns, methods such as Naïve Bayes might be of value.

Robustness of machine learning methods to different levels of noise for 4 out of 7 activity classes. At low levels of noise, lower that 20%, non-linear methods performed well achieving performance higher than 0.7 MCC units for most of the tested datasets. Instead, at higher level of noise, equal to or higher than 30%, performance for most algorithms dropped below 0.7 MCC and in several occasions even lower than 0.6 at 50% of noise. Naïve Bayes method was found to be the least affected method achieving in several tested datasets performance higher than 0.6 MCC even at the highest level of noise tested 50% and outperforming more complex methods
Conclusions
In this study feed-forward deep neural networks were investigated for modeling bioactivity data. The aim of the study was dual, first DNN hyper-parameters values were explored to investigate how they affect the performance of feed-forward DNN. Below we summarize and compare our findings with those reported by Ma et al. [19], where strong agreement were observed:
-
Rectified linear units (ReLU) activation function performed overall better than Sigmoid or Tanh. Hence, it is highly recommended to use ReLU activation function when training DNN.
-
The number of hidden layers should be at least 2 or 3 in order for DNN to achieve strong performance. Further increase of number of hidden layers had diminishing returns as best results were obtained when using 2 or 3 hidden layers.
-
The number of neurons per hidden layer was found to depend on the dataset and should be optimized on a case-by-case basis. While it’s recommended to use at least 100 neurons in each hidden layer, best results were obtained when using larger number of neurons, e.g. 700, 1000, 2000 or higher.
-
Dropout regularization technique was found to have great impact on the performance of DNN by preventing co-adaption of neurons. Here it was found that dropout should be applied to both input and hidden layers. Good performance was achieved when dropout of 50% was applied to both input and hidden layers.
-
No pre-training of DNN was performed and both the weights and biases were randomly initialized from Gaussian distribution. Similarly, Ma et al. reported that no unsupervised pre-training is needed and network parameters should be initialized as random values.
-
The number of epochs should be as many as possible, at least 300, given the local computing capability. Modern GPU units provide adequate computational power to optimizing DNN configurations in reasonable time. Combined with Dropout technique over-fitting shouldn’t be a major concern.
In the second part of the study, the performance of DNNs was compared to widely popular shallow methods Bernoulli Naïve Bayes, k-nearest neighbor, random forest and support vector machines. Here it was found that optimized deep neural nets achieved statistically better performance, measured by the Wilcoxon test on the tested datasets. These results demonstrate the deep-learning techniques can serve as powerful modeling techniques for modeling complex biological data. While deep learning techniques are highly unlikely to replace existing algorithms any time soon, their popularity in the field of Cheminformatics is expected to grow as the availability of GPU hardware and of stable and well-documented software packages become more accessible to Cheminformatic practitioners.
Interpretation of local and global SAR captured by complex QSAR models remains a challenging task, in particular when nonlinear algorithms are employed [45]. Several approaches in the literature have been proposed for interpreting local and global QSAR models based on support vector machine and random forest techniques [46, 47]. DNNs are generally considered as “black boxes” and are difficult to be interpreted [15]. Hence, future studies should aim to investigate how models learned by DNN could be interpreted, as this could help rationalizing structure–activity relationships encoded by models. This knowledge could drive the lead optimization process by suggesting structural modifications that might be beneficial for the primary activity, while decreasing any undesired off-target interactions.
Methods and Materials
Bioactivity datasets
As source of structure–activity relationships (SARs) the ChEMBL database was employed [48, 49]. ChEMBL, version 20, was used in this study. In total seven diverse bioactivity classes were selected and used in the study: (a) Carbonic Anhydrase II (ChEMBL205), a protein lyase, (b) Cyclin-dependent kinase 2 (CHEMBL301), a protein kinase, (c) ether-a-go-go-related gene potassium channel 1 (HERG) (CHEMBL240), a voltage-gated ion channel, (d) Dopamine D4 receptor (CHEMBL219), a monoamine GPCR, (e) Coagulation factor X (CHEMBL244), a serine protease, (f) Cannabinoid CB1 receptor (CHEMBL218), a lipid-like GPCR and (g) Cytochrome P450 19A1 (CHEMBL1978), a cytochrome P450. The activity classes were selected based on data availability and as representatives of therapeutically important target classes or as anti-targets.
SARs data were extracted following the criteria: (a) Only human direct protein targets were considered, (b) confidence score equal to 9, (c) MW up to 900, (d) activity was defined based on pKi or pIC50, depending which type of measurement was the majority of available per activity class, (e) active compounds were defined those with potency, pKi or pIC50, better than or equal to 5, which equals to or better than 10 μM. Decoys were randomly sampled from a large pool of drug-like compounds extracted from ChEMBL, covering small bioactive molecules reported against protein targets with potencies (Ki/IC50/Kd) up to 10 μM and MW up to 900. As an additional step chemical structures with Tanimoto similarity coefficient larger than 0.9 were removed. For each active structure, 10 decoys were randomly sampled. The main motivation here was to assemble reasonably sized datasets for each activity class, suitable for comparing the performance of the machine learning methods included in the study while avoiding creating highly unbalanced datasets. Number of active and decoys assembled per activity class is shown in Table 1.
Chemical structures were standardized using the MOE software package with the options on: (a) disconnect group/metals in simple salts, (b) keep only largest molecular fragments, (c) deprotonate strong acids, (d) protonate strong bases and (e) replace coordinates with a generated 2D depiction [50]. As molecular descriptors the Morgan fingerprints as implemented in the Rdkit Cheminformatic toolkit were utilized with radius 2, which are equal to extended connectivity fingerprints (ECFP_4), and stored in hashed 1024 bit length binary vectors [51, 52]. ECFP fingerprints were selected for this study as they have been previously shown to perform well and are commonly employed in virtual screening applications [53]. The datasets utilized in this study are provided in Additional file 1.
Machine learning methods
Bernoulli Naïve Bayes
The first of the shallow algorithms employed in this study was the Bernoulli Naïve Bayes classifier (NB) [54]. Naïve Bayes classifier is a probabilistic supervised machine-learning algorithm based on the Bayes’ theorem with the strong “naive” assumption of feature independence [54,55,56]. NB was included as a baseline method in this study as implemented in the Scikit-learn library utilizing the “BernoulliNB” function [57]. The explored hyper-parameters values are presented in Table 2.
k-Nearest neighbor
The second algorithm employed was the k-nearest neighbor (kNN) [58]. K-Nearest Neighbor was utilized as implemented in the Scikit-learn library. Here the “KNeighborsClassifier” function was employed. Only the hyper-parameter number of nearest neighbors was explored while the rest were retained default. The explored hyper-parameters values are presented in Table 2.
Random forest
The third algorithm employed was the random forest (RF). RF is an ensemble of multiple weak un-pruned classification or regression trees created by using bootstrap samples of the training data and random feature selection in tree induction [59]. RF was developed by Breiman and Cutler [60] and presents a number of advantages, which makes it attractive and well suited for cheminformatic applications; (a) it is robust when dealing with large number of features, (b) achieves generally good performance, (c) resilient to over-fitting and (d) can be parallelized across multiple CPU cores. Furthermore, it can be employed to assess the relevance of features, effectively acting as a feature selection algorithm [61, 62]. RF as implemented in the scikit-learn library was employed, “RandomForestClassifier” function from the ensemble family [57]. The explored hyper-parameters values are presented in Table 2.
Support vector machines
The fourth algorithm employed was the support vector machines (SVMs). SVMs along with RF is a widely applied machine-learning algorithm in the field of Cheminformatics [63, 64]. SVMs was proposed by Cortes and Vapnik and can be applied for classification, regression and outliers detection tasks [65]. In this study, two kernels were considered the non-linear radial-basis function and the linear kernel. SVM as implemented in the scikit-learn library was employed. Scikit-learn implementation of SVM is based on the Libsvm and the Liblinear libraries [66, 67]. When optimizing the SVM using the non-linear RBF kernel the values for hyper-parameters gamma (γ) and Cost (C) where selected with similar range to those reported by Alvarsson et al. [68]. Here values for gamma (γ) tested were 10e−1, 10e−2, 10e−3, 10e−4, 10e−5 and for cost (C) 1, 10, 100, 1000. The explored hyper-parameters values are presented in Table 2. Results obtained when optimizing the SVM algorithm using the RBF kernel were in good agreement with those reported by Alvarsson et al., where for gamma (γ) equal to 0.01 and Cost (C) values of 10, 100 and 1000 the highest values in performance were obtained across all datasets.
Deep neural networks
As mentioned earlier the Caffe deep learning framework was adopted in this study. In order to automate the process and investigate multiple hyper-parameters configurations the Python wrapper “ChemoCaffe_tune.py” and “ChemoCaffe_predict.py” were developed, which are built upon the pyCaffe module. Caffe requires the input data to be either in LMDB [69] or HDF5 [70] format; we opted for the HDF5 format mainly due to convenience. No feature transformation or pre-training of DNNs was employed in this study. DNNs were optimized on a dedicated server with NVIDIA Tesla K40 GPU units.
The hyper-parameters selected and explored for DNNs were: (A) Default parameters, those that were kept fixed across all tested configurations: (a) As optimization method the Stochastic Gradient Descent (“SGD”) with momentum μ = 0.9 was used, (b) number of epochs was set to 300, (c) mini-batch size of 256, (d) Weights and biases were initialized from Gaussian distribution with Standard deviation of 0.01 and (e) As loss function the logistic softmax was used, “SoftmaxWithLoss” and (d) softmax as the output function for calculating class probabilities and (g) the weight_decay was set to 0.0005. (B) Variable hyper-parameters: (a) Activation functions compared the rectified linear units (ReLU), Sigmoid (“sigm”) and Tanh (“tanh”), Fig. 1c illustrates the shapes of the functions. (b) Number of neurons in each hidden layer (5, 10, 50, 100, 200, 500, 700, 1000, 1500, 2000, 2500, 3000, 3500), (c) learning rate “η” of (1, 10−1, 10−2, 10−3, 10−4), (d) number of hidden layers up to 4, (e) regularization technique applied; (1) no regularization and (2) dropout. In case of dropout regularization: for input layer dropout of 0, 20 and 50% were applied and for the hidden layer of 50%. The explored hyper-parameters values are presented in Table 2. The python wrappers for tuning hyper-parameters “ChemoCaffe_tune.py” and generating predictions “ChemoCaffe_predict.py” used to carry out this study as also the configuration files containing the tested configurations are provided in Additional file 2.
Performance metric
Matthews correlation coefficient (MCC)
As performance evaluation metric the Matthews correlation coefficient (MCC) was utilized to measure and compare the performance of machine learning algorithms employed in the study [71, 72]. MCC is a commonly applied performance evaluation metric to measure the quality of binary classification and can be calculated according to the Eq. (1). It measures the correlation between predicted and the actual class labels. MCC is generally considered to be a balanced evaluation metric and takes values between −1 and +1, where −1 indicates perfect anti-correlation between predicted and real observations, +1 represents perfect prediction and 0 that equals to no better than random,
where TP are true positives, TN true negatives, FP false positives and FN false negatives (Additional file 3).
Wilcoxon signed-rank test
Wilcoxon paired singed-rank test a non-parametric statistical hypothesis test was used to compare matched samples to assess whether their population mean ranks differ [73]. Wilcoxon test as implemented in the R programming language was utilized, part of the “stats” package, with confidence interval 99% and alternative hypothesis being “greater” [74].
References
Bains W, Gilbert R, Sviridenko L, Gascon JM, Scoffin R, Birchall K, Harvey I, Caldwell J (2002) Evolutionary computational methods to predict oral bioavailability QSPRs. Curr Opin Drug Discov Dev 5:44–51
Basak SC, Mills D (2001) Quantitative structure–property relationships (QSPRs) for the estimation of vapor pressure: a hierarchical approach using mathematical structural descriptors. J Chem Inf Model 41:692–701
Le T, Epa VC, Burden FR, Winkler DA (2012) Quantitative structure–property relationship modeling of diverse materials properties. Chem Rev 112:2889–2919
Cronin MTD (2010) Quantitative structure–activity relationships (QSARs)—applications and methodology. In: Puzyn T, Leszczynski J, Cronin MT (eds) Recent advances in QSAR studies methods and applications. Springer, pp 3–11
Kruhlak NL, Benz RD, Zhou H, Colatsky TJ (2012) (Q)SAR modeling and safety assessment in regulatory review. Clin Pharmacol Ther 91:529–534
Merlot C (2010) Computational toxicology—a tool for early safety evaluation. Drug Discov Today 15:16–22
Koutsoukas A, Simms B, Kirchmair J, Bond PJ, Whitmore AV, Zimmer S, Young MP, Jenkins JL, Glick M, Glen RC, Bender A (2011) From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics 74:2554–2574
Winkler DA (2004) Neural networks as robust tools in drug lead discovery and development. Mol Biotechnol 27:139–168
Mosier PD, Jurs PC (2002) QSAR/QSPR studies using probabilistic neural networks and generalized regression neural networks. J Chem Inf Model 42:1460–1470
Burden FR (1999) Robust QSAR models using bayesian regularized neural networks. J Med Chem 42:3183–3187
Huuskonen J, Salo M, Taskinen J (1998) Aqueous solubility prediction of drugs based on molecular topology and neural network modeling. J Chem Inf Comput Sci 38:450–456
Lucic B, Trinajstic N (1999) Multivariate regression outperforms several robust architectures of neural networks in QSAR modeling. J Chem Inf Model 39:121–132
Niculescu SP (2003) Artificial neural networks and genetic algorithms in QSAR. J Mol Struct (Thoechem) 622:71–83
Agrafiotis DK, Cedeno W, Lobanov VS (2002) On the use of neural network ensembles in QSAR and QSPR. J Chem Inf Model 42:903–911
Guha R, Jurs PC (2005) Interpreting computational neural network QSAR models: a measure of descriptor importance. J Chem Inf Model 45:800–806
Manallack DT, Livingstone DJ (1999) Neural networks in drug discovery: have they lived up to their promise? Eur J Med Chem 34:195–208
Schneider G (2000) Neural networks are useful tools for drug design. Neural Netw 13:15–16
Gawehn E, Hiss JA, Schneider G (2016) Deep learning in drug discovery. Mol Inform 35:3–14
Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V (2015) Deep neural nets as a method for quantitative structure–activity relationships. J Chem Inf Model 55:263–274
Andreas Mayr GK, Unterthiner T, Hochreiter S (2016) DeepTox: toxicity prediction using deep learning. Front Environ Sci 3:80
Arel I, Rose DC, Karnowski TP (2010) Deep machine learning—a new frontier in artificial intelligence research [research frontier]. IEEE Comput Intell Mag 5:13–18
LeCun Y, Bengio Y, Hinton G (2015) Deep learning. Nature 521:436–444
Bengio Y, Courville A, Vincent P (2013) Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell 35:1798–1828
Dahl G, Mohamed A-R, Hinton GE (2010) Phone recognition with the mean-covariance restricted Boltzmann machine. Adv Neural Inf Process Syst 2010:469–477
Ciresan D, Meier U, Schmidhuber J (2012) Multi-column deep neural networks for image classification. In: Proceedings of the IEEE international conference on computer vision and pattern recognition (CVPR), pp 3642–3649
Cireşan DC, Meier U, Gambardella LM, Schmidhuber J (2010) Deep, big, simple neural nets for handwritten digit recognition. Neural Comput 22:3207–3220
Basheer IA, Hajmeer M (2000) Artificial neural networks: fundamentals, computing, design, and application. J Microbiol Methods 43:3–31
McCulloch WS, Pitts W (1943) A logical calculus of the ideas immanent in nervous activity. Bull Math Biophys 5:115–133
Larochelle H, Bengio Y, Louradour M, Lamblin P (2009) Exploring strategies for training deep neural networks. J Mach Learn Res 10:1–40
Rumelhart DE, Hinton GE, Williams RJ (1986) Learning representations by back-propagating errors. Nature 323:533–536
Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R (2014) Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res 15:1929–1958
Owens JD, Houston M, Luebke D, Green S, Stone JE, Phillips JC (2008) GPU computing. Proc IEEE 96:879–899
Lusci A, Pollastri G, Baldi P (2013) Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model 53:1563–1575
Xu Y, Dai Z, Chen F, Gao S, Pei J, Lai L (2015) Deep learning for drug-induced liver injury. J Chem Inf Model 55:2085–2093
Aliper A, Plis S, Artemov A, Ulloa A, Mamoshina P, Zhavoronkov A (2016) Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm 13:2524–2530
Aspuru-Guzik A, Duvenaud D, Maclaurin D, Aguilera-Iparraguire J, Gomez-Bombarelli R, Hirzel TD, Adams RP (2015) Convolutional networks on graphs for learning molecular fingerprints. In: Proceedings of neural information processing systems, Quebec, pp 2224–2232
Team TTD, Al-Rfou R, Alain G, Almahairi A, Angermueller C, Bahdanau D, Ballas N, Bastien F, Bayer J, Belikov A (2016) Theano: a Python framework for fast computation of mathematical expressions. arXiv preprint arXiv:1605.02688
Jia Y, Shelhamer E, Donahue J, Karayev S, Long J, Girshick R, Guadarrama S, Darrell T (2014) Caffe: convolutional architecture for fast feature embedding. In: Proceedings of the ACM international conference on multimedia, 2014. ACM, pp 675–678
Zeiler MD, Ranzato M, Monga R, Mao M, Yang K, Le QV, Nguyen P, Senior A, Vanhoucke V, Dean J, Hinton GE (2013) On rectified linear units for speech processing. In: Proceedings of IEEE international conference on acoustics, speech and signal processing, Vancouver, pp 3517–3521
Tiikkainen P, Franke L (2012) Analysis of commercial and public bioactivity databases. J Chem Inf Model 52:319–326
Williams AJ, Ekins S, Tkachenko V (2012) Towards a gold standard: regarding quality in public domain chemistry databases and approaches to improving the situation. Drug Discov Today 17:685–701
Kramer C, Kalliokoski T, Gedeck P, Vulpetti A (2012) The experimental uncertainty of heterogeneous PublicKiData. J Med Chem 55:5165–5173
Kramer C, Dahl G, Tyrchan C, Ulander J (2016) A comprehensive company database analysis of biological assay variability. Drug Discov Today 21:1213–1221
Glick M (2004) Enrichment of extremely noisy high-throughput screening data using a Naive Bayes classifier. J Biomol Screen 9:32–36
Guha R (2008) On the interpretation and interpretability of quantitative structure–activity relationship models. J Comput Aided Mol Des 22:857–871
Carlsson L, Helgee EA, Boyer S (2009) Interpretation of nonlinear QSAR models applied to AMES mutagenicity data. J Chem Inf Model 49:2551–2558
Rosenbaum L, Hinselmann G, Jahn A, Zell A (2011) Interpreting linear support vector machine models with heat map molecule coloring. J Cheminform 3:11
Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, Krüger FA, Light Y, Mak L, McGlinchey S, Nowotka M, Papadatos G, Santos R, Overington JP (2014) The ChEMBL bioactivity database: an update. Nucleic Acids Res 42:D1083–D1090
Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, Overington JP (2011) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40:D1100–D1107
Molecular Operating Environment (MOE) 2013.08. Chemical Computing Group Inc.: Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2016
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742–754
Landrum G. RDKit: Open-source cheminformatics. http://www.rdkit.org
Bender A, Jenkins JL, Scheiber J, Sukuru SCK, Glick M, Davies JW (2009) How similar are similarity searching methods? A principal component analysis of molecular descriptor space. J Chem Inf Model 49:108–119
McCallum A, Nigam K (1998) A comparison of event models for Naive Bayes text classification. In: Proceedings of the AAAI-98 workshop on learning for text categorization, vol 752, pp 41–48
Lewis DD (1998) Naive (Bayes) at forty: the independence assumption in information retrieval. In: Nédellec C, Rouveirol C (eds) Machine learning: ECML-98. ECML 1998. Lecture notes in computer science, vol 1398. Springer, Berlin, Heidelberg, pp 4–15
Jordan A (2002) On discriminative vs. generative classifiers: a comparison of logistic regression and naive bayes. Adv Neural Inf Process Syst 14:841
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V (2011) Scikit-learn: machine learning in Python. J Mach Learn Res 12:2825–2830
Altman NS (1992) An introduction to kernel and nearest-neighbor nonparametric regression. Am Stat 46:175–185
Svetnik V, Liaw A, Tong C, Culberson JC, Sheridan RP, Feuston BP (2003) Random forest: a classification and regression tool for compound classification and QSAR modeling. J Chem Inf Model 43:1947–1958
Breiman L (2001) Random forests. Mach Learn 45:5–32
Saeys Y, Inza I, Larranaga P (2007) A review of feature selection techniques in bioinformatics. Bioinformatics 23:2507–2517
Díaz-Uriarte R, Alvarez de Andrés S (2006) Gene selection and classification of microarray data using random forest. BMC Bioinform 7:3
Mitchell JBO (2014) Machine learning methods in chemoinformatics. Wiley Interdiscipl Rev Comput Mol Sci 4:468–481
Barrett SJ, Langdon WB (2006) Advances in the application of machine learning techniques in drug discovery. Des Dev 36:99–110
Cortes C, Vapnik V (1995) Support-vector networks. Mach Learn 20:273–297
Chang C-C, Lin C-J (2011) Libsvm. ACM Trans Intell Syst Technol 2:1–27
Fan R-E, Chang K-W, Hsieh C-J, Wang X-R, Lin C-J (2008) LIBLINEAR: a library for large linear classification. J Mach Learn Res 9:1871–1874
Alvarsson J, Eklund M, Andersson C, Carlsson L, Spjuth O, Wikberg JE (2014) Benchmarking study of parameter variation when using signature fingerprints together with support vector machines. J Chem Inf Model 54:3211–3217
Chu H. OpenLDAP lighting memory-mapped database. http://symas.com/mdb/
Group TH. Hierarchical data format (HDF). www.hdfgroup.org
Matthews BW (1975) Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta (BBA) Protein Struct 405:442–451
Baldi P, Brunak S, Chauvin Y, Andersen CAF, Nielsen H (2000) Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics 16:412–424
Wilcoxon F (1945) Individual comparisons by ranking methods. Biom Bull 1:80
Team RC (2014) In: ISBN 3-900051-07-0
Authors’ contributions
AK, KJM, XL and JH designed the experiments. AK and KJM extracted the datasets and wrote the code used in the study. AK and KJM run the experiments. AK, KJM, XL and JH wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors thank the Advanced Computing Facility of the University of Kansas (https://corelabs.ku.edu/advanced-computing-facility) for the support throughout this research. We thank Dr. Min Shen at National Center for Advancing Translational Sciences for her help in performing research on applying deep learning for cheminformatics.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The seven activity classes extracted from ChEMBL database and used in this study are provided in the Additional file 4 free of charge. The scripts used to generate circular fingerprints based on RDkit toolkit, split datasets for parameter tuning/training and validation sets are made publicly available. The scripts used to tune each algorithm RF, SVM, kNN, Naïve Bayes based on the scikit-learn library and ChemoCaffe based on Caffe framework are made publicly available.
Funding
This Project was funded by NSF Grant CNS 1337899. We gratefully acknowledge the support of NVIDIA Corporation with the donation of the Tesla K40 GPUs used for this research.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Koutsoukas, A., Monaghan, K.J., Li, X. et al. Deep-learning: investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J Cheminform 9, 42 (2017). https://doi.org/10.1186/s13321-017-0226-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-017-0226-y