
Abstract
Background
DNA methylation is associated with cancer, metabolic, neurological, and autoimmune disorders. Hypomethylation of aryl hydrocarbon receptor repressor (AHRR) especially at cg05575921 is associated with smoking and lung cancer. Studies on the association between AHRR methylation at cg05575921 and sources of polycyclic aromatic hydrocarbon (PAH) other than smoking are limited. The aim of our study was to assess the pattern of blood DNA methylation at cg05575921 in non-smoking Taiwanese adults living in areas with different PM2.5 levels.
Methods
Data on blood DNA methylation, smoking, and residence were retrieved from the Taiwan Biobank dataset (2008–2015). Current and former smokers, as well as individuals with incomplete information were excluded from the current study. The final analysis included 708 participants (279 men and 429 women) aged 30–70 years. PM2.5 levels have been shown to increase as one moves from the northern through central towards southern Taiwan. Based on this trend, the study areas were categorized into northern, north-central, central, and southern regions.
Results
Living in PM2.5 areas was associated with lower methylation levels: compared with the northern area (reference area), living in north-central, central, and southern areas was associated with lower methylation levels at cg05575921. However, only methylation levels in those living in central and southern areas were significant (β = − 0.01003, P = 0.009 and β = − 0.01480, P < 0.001, respectively. Even though methylation levels in those living in the north-central area were not statistically significant, the test for linear trend was significant (P < 0.001). When PM2.5 was included in the regression model, a unit increase in PM2.5 was associated with 0.00115 (P < 0.001) lower cg05575921 methylation levels.
Conclusion
Living in PM2.5 areas was inversely associated with blood AHRR methylation levels at cg05575921. The methylation levels were lowest in participants residing in southern followed by central and north-central areas. Moreover, when PM2.5 was included in the regression model, it was inversely associated with methylation levels at cg05575921. Blood methylation at cg05575921 (AHRR) in non-smokers might indicate different exposures to PM2.5 and lung cancer which is a PM2.5-related disease.
Similar content being viewed by others

Background
DNA methylation, an epigenetic process characterized by the addition of a methyl group to a DNA molecule influences the expression of genes [1, 2]. It is associated with several health conditions including cancer, metabolic, neurological, psychiatric, and autoimmune disorders [1,2,3,4,5]. A greater portion of DNA methylation in humans takes place in regions called CG or CpG sites, where a cytosine precedes a guanine nucleotide [1, 2]. Due to its epigenetic nature, DNA methylation is associated with several environmental factors including cigarette smoking, exercise, and alcohol drinking, among others [6, 7].
The gene, aryl hydrocarbon receptor repressor (AHRR), mediates the metabolism of xenobiotic particles like toxic cigarette smoke components [8, 9]. This gene is located on chromosome 5 which is believed to possess several tumor suppressor genes [10, 11]. Blood AHRR methylation (especially at cg0557592) is inversely associated with both smoking [4, 5, 8, 9, 12,13,14,15,16] and lung cancer [4, 12, 13].
Cigarette smoke and PM2.5 are both inhalable carcinogenic factors composed of a complex mixture of chemicals, one of which is polycyclic aromatic hydrocarbons (PAHs) [11, 17,18,19]. PM-related PAHs pose most of the health problems in humans [20]. Exposure to PAHs is capable of activating the aryl hydrocarbon receptor (AHR) [17, 21] and is thought to be responsible for variations in smoking-related AHRR methylation [9]. Even though AHR activation is associated with particulate matter [22, 23], research on the association between methylation at cg05575921 (AHRR) and PM2.5 is limited. In a cross-sectional study, AHRR (cg05575921) hypomethylation in non-smokers was associated with exposure to second-hand smoke (SHS) but not PM2.5 [24]. Moreover, in a recent review on DNA methylation and environmental factors, several studies were listed to have demonstrated associations between AHRR hypomethylation and tobacco smoke. However, none was listed to have demonstrated an association between AHRR hypomethylation and exposure to air pollution or PAHs [25].
In a study conducted in Taiwan, PM2.5 was used as a proxy marker for PAHs [20]. PM2.5 concentrations in northern Taiwan are lower than in central and southern areas [26,27,28,29]. Because PAHs are present in both PM2.5 and cigarette smoke, AHRR (cg05575921) methylation in smokers might be comparable with that in non-smokers exposed to PM2.5. Therefore, we used an epigenetic approach to investigate the pattern of blood methylation at cg05575921 in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations.
Results
Table 1 shows the general characteristics of study participants. There were 708 participants comprising 279 men (mean age = 49.42 ± 11.76 years) and 429 women (mean age = 49.49 ± 10.97 years). The mean (± SD) methylation level at cg05575921 was 0.83604 ± 0.00154. In general, 244, 121, 134, and 209 participants resided in the northern, north-central, central, and southern areas, respectively. Table 2 shows the mean (± SD) concentrations of PM2.5 (μg/m3) from 2006 to 2011 in the study areas. These concentrations were 27.32 ± 4.34, 28.65 ± 2.13, 35.72 ± 3.75, and 39.81 ± 2.10 μg/m3 for the northern, north-central, central, and southern areas, respectively. Table 3 shows the association between living in PM2.5 areas and AHRR (cg05575921) methylation. With the northern area as the reference, residing in north-central, central, and southern areas was associated with lower blood methylation levels at cg05575921. When SHS was included in the analysis, the regression coefficients (β) were − 0.00274 (P = 0.503), − 0.01003 (P = 0.009), and − 0.01480 (P < 0.001), respectively (Table 3, model 1). That is, the blood methylation levels in participants residing in north-central, central, and southern areas were lower when compared to those in the northern area. The differences were − 0.00274, − 0.01003, and − 0.01480, respectively. After SHS was excluded from the analysis, the regression coefficients (β) were − 0.00028 (P = 0.947), − 0.01069 (P = 0.009), and − 0.01487 (P < 0.001), respectively (Table 3, model 2). Even though methylation levels in participants who lived in north-central areas were not statistically significant, the test for linear trend was statistically significant (P trend < 0.001) in both models (Table 3, models 1 and 2). The mean PM2.5 concentration from 2006–2011 was significantly associated with lower blood AHRR methylation levels at cg05575921 (Table 4). A unit increase in PM2.5 concentration was associated with 0.00115 (P < 0.001) lower methylation when SHS was included in the analysis (Table 4, model 1). Similarly, a unit increase in PM2.5 concentration was associated with 0.00124 (P < 0.001) lower methylation after SHS was excluded from the analysis (Table 4, model 2). Spearman analysis showed a significant negative correlation (β = − 0.78329; P < 0.001) between PM2.5 concentration (μg/m3) and mean methylation levels (Additional file 1). The associations between PM2.5 and 176 sites in the AHRR promoter region are shown in Additional file 2. In addition to cg05575921, some other sites that were significantly associated with PM2.5 include cg26703534 (β = − 0.00127; P < 0.001), cg25648203 (β = − 0.00078; P < 0.001), and cg21161138 (β = − 0.00046; P = 0.007).
Discussion
Studies on the association between AHRR methylation and sources of polycyclic aromatic hydrocarbon (PAH) other than smoking are limited. To our knowledge, the current study is the first to assess blood AHRR methylation in non-smokers residing in areas with different concentrations of PM2.5. In general, PM2.5 was inversely associated with blood AHRR (cg05575921) methylation. Compared with the northern area, living in north-central, central, and southern areas was associated with lower blood AHRR methylation levels at cg05575921.
Living in areas with higher PM2.5 concentrations has been associated with greater exposure to PAH [20]. Particulate matter-related PAHs have been reported as potential activators of AHR [17]. PM2.5 and cigarette smoke both contain PAHs which can induce AHRR methylation [11]. Moreover, they are both inhalable and carcinogenic [19]. Several studies have explored the association between PM2.5 and DNA methylation in blood [23, 30] and placenta [31]. Nonetheless, emphasis has not been laid on blood AHRR methylation in non-smokers. In light of this, we investigated the association between living in PM2.5 areas and AHRR methylation at cg05575921 in non-smokers. We adjusted for exposure to SHS since significant inverse associations have been found between methylation at cg05575921 and exposure to SHS in non-smokers [24].
Cg05575921 was selected because it has been shown to be highly sensitive and specific in identifying and distinguishing smoking status as well as tracking trends in smoking termination and reduction [12, 15, 32,33,34]. For instance, current smokers showed the highest hypomethylation compared to former and or non-smokers [4, 5, 13,14,15,16]. Moreover, this methylation site has been shown to be the most statistically significant smoking-related site in several original investigations [13, 16, 35,36,37,38] and a meta-analysis [39].
In our study, it was hypothesized that blood methylation patterns at cg05575921 (AHRR) in non-smokers residing in areas with different PM2.5 concentrations might be similar to those observed based on smoking status. As expected, the results were similar to those from several studies that were conducted among non-, former, and current smokers [4, 5, 13,14,15,16]. That is, blood methylation levels at cg05575921 in non-smokers residing in Southern Taiwan were lowest followed by Central and Northern Taiwan with significant linear trends. The regression coefficients were − 0.00028, − 0.01069, and − 0.01487 for north-central, central, and southern areas, respectively. Previous studies have reported regression coefficients of 0.83, 0.79, and 0.59 for never, former, and current smokers [5] and 0.878, 0.829, and 0.772 for non-, light, and heavy smokers, respectively [16]. Moreover, the methylation extent was 64, 60, and 50 % among never, former, and current smokers [4]. PM2.5 levels are higher in Southern and Central compared to Northern Taiwan [26,27,28,29]. There are many heavy industries (e.g., petrochemical plants) in these areas [27, 28, 40]. Emissions from these industries are believed to be one of the main sources of PM2.5- and PM2.5-bound PAHs [41,42,43]. Higher concentrations of PM2.5-bound PAHs have also been reported in the industrial cities of Christchurch and Guadalajara in New Zealand [43, 44] and Mexico [43, 45].
Some of the merits of our study include (1) the relatively larger sample size, (2) the adjustment for exposure to SHS to avoid its confounding effect, and (3) stratification of participants into four areas known for different PM2.5 levels. However, the study is limited in that DNA methylation was determined using the Illumina Infinium MethylationEPIC BeadChip which covers 850,000 methylation sites. Even though this microarray has been validated as a very reliable genomic platform for determining DNA methylation patterns in the human genome [46], validation experiments were not performed to confirm our findings. Therefore, future investigations to support our results are recommended. Moreover, functional correlation between cg05575921 methylation and AHRR mRNA gene expression was not evaluated in this study. Furthermore, the actual concentrations of PM2.5 in individuals could not be determined since there are no validated tools for individual exposure estimates. Air quality indices from nearby monitoring stations are usually used for air pollution estimation [47]. In the current study, the number of monitoring stations corresponding to the participants’ residence was relatively small. This is because some counties involved in the study do not have monitoring stations; hence, PM2.5 estimates were not available for participants living in such areas. However, we think our results are plausible because variations in PM2.5 concentration in various areas in Taiwan are well known [26,27,28,29]. Moreover, the inclusion of mean PM2.5 concentration from the study areas (2006–2011) into our regression model showed significant inverse associations between PM2.5 and blood methylation at cg05575921 (AHRR). The fact that other environmental factors would have also accounted for this association cannot be completely rolled out. However, the emphasis on PM2.5 is because it contains PAHs which are related to AHRR. Exposure to PAHs is believed to cause smoking-related AHRR methylation [9]. Moreover, PM2.5 is a serious problem in Taiwan and PM-related PAHs pose most of the health problems in humans [20].
Conclusion
In conclusion, living in PM2.5 areas was inversely associated with blood AHRR methylation levels at cg05575921. Compared with Northern Taiwan (which has the lowest PM2.5 levels), living in North-Central, Central, and Southern Taiwan was associated with lower blood AHRR methylation levels at cg05575921. The methylation levels were lowest in those residing in the southern area (which has the highest PM2.5) levels followed by central and north-central areas. Moreover, PM2.5 was inversely associated with methylation levels at cg05575921 when it was included in the regression model. AHRR (cg05575921) methylation might serve as an indicator of differential exposures to PM2.5 and lung cancer which is also a PM2.5-related disease. Further investigations to confirm these findings are recommended.
Methods
Data source
Data used in the current study were obtained from the Taiwan Biobank which was founded in 2005. The biobank aims at undertaking large-scale cohort and case-control studies through the combination of genetic and medical information [48, 49]. The information collected is currently serving as one of the cornerstones of medical research in Taiwan. It is hoped that the identification of disease risk factors and the underlying mechanisms would lead to the development of better treatments and prevention strategies, hence, reduced medical costs [48, 49]. As a result, the health of the population would probably be promoted and improved.
Information about recruitment in the Taiwan Biobank project is obtained through brochures, posters, media, and websites. Interested volunteers are asked to provide their contact information. These volunteers are contacted to confirm their willingness to participate, and those who fulfill the recruitment requirements (strictly Taiwanese aged 30–70 years with no personal history of cancer) are assigned to specific recruitment centers. Currently, the biobank comprises 29 recruitment centers with each city or county having at least 1 center [49]. Before data are collected, all the participants sign a letter of consent.
Data collection
Data were collected by trained medical researchers through questionnaires (e.g., residence, sex, age, smoking, alcohol drinking, exercise habits, and exposure to SHS), physical examination (e.g., weight and height), and blood examination (e.g., DNA methylation). Participants were considered as non-smokers if they reported to have never smoked or have not continuously smoked for at least 6 months. Former smokers were those who reported to have continuously smoked for at least 6 months but were currently not smoking. Current smokers were those who reported to have continuously smoked for at least 6 months and were currently smoking. Participants were considered non-drinkers if they did not drink or drank less than 150 cc of alcohol per week continuously for 6 months. Former drinkers were those who abstained from alcohol for more than 6 months while current drinkers were those who drank at least 150 cc of alcohol per week continuously for 6 months. Exercise habits were categorized under “yes” if participants reported a habit of exercising at least three times per week (each exercise time > 30 min) and under “no” if they did not exercise at least three times per week. Individuals considered to be exposed to SHS were those who reported being exposed to SHS for at least 5 minutes per hour. BMI was derived from weight and height as BMI = weight (kg)/height (m2).
DNA was extracted from whole blood using an automated extraction machine called chemagicTM PrimeTM instrument. DNA length was checked using Fragment Analyzer (Agilent). Its quality was assessed using the ratio of absorbance at 260/280 with the purity index set as 1.6–2.0. The samples that passed quality control were stored at − 80 °C for long-term use. DNA methylation was determined using the Illumina Infinium MethylationEPIC BeadChip which has been previously described [50,51,52]. In brief, DNA samples were treated with sodium bisulfite conversion using the EZ DNA Methylation Kit (Zymo Research, CA, USA). Data quality control was performed based on the Illumina® GenomeStudio® Methylation Module v1.8 [53]. Samples with P value > 0.05 or bead count < 3 were removed. Dye bias across batches was adjusted by normalization and background correction was performed. Outliers were removed using the median absolute deviation method. The methylation level at each CpG site was determined using β values. The β values were derived using the formula β = M/(M + U), where M = methylated intensity and U = unmethylated intensity.
Study participants
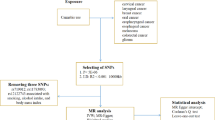
Data used in the current study were retrieved from the Taiwan Biobank dataset (2008–2015). A total of 1142 individuals aged between 30 and 70 years with no personal history of cancer were initially enrolled. We excluded 301 current and former smokers, as well as 133 individuals who did not live in the study area (Fig. 1) from the study. The final sample included 708 non-smoking participants consisting of 279 men and 429 women.

Map showing the study areas and the location of monitoring stations
The study area was categorized into Northern (Taipei and New Taipei Cities), North-Central (Taoyuan and Hsinchu Cities, Hsinchu, Miaoli, and Taoyuan Counties), Central (Taichung City, Nantou, Changhua, and Yunlin Counties), and Southern Taiwan (Chiayi County, Tainan, and Chiayi Cities). These areas are well known for varying PM2.5 levels. That is, PM2.5 levels increase as one moves from the north through the center towards the south of Taiwan [26,27,28,29]. Overall, there were 244, 121, 134, and 209 participants residing in the northern, north-central, central, and southern areas, respectively. The number of monitoring stations was 18 for the northern, 12 for the north-central, 16 for the central, and 7 for the southern area. In this study, we used the average PM2.5 levels (2006–2011) of the monitors of each area (northern, north-central, central, and southern areas) to estimate the PM2.5 exposure of participants. The location of participants and monitoring stations is shown in Fig. 1. The current study was approved by the Chung Shan Medical University Institutional Review Board (CS2-17070).
Statistical analysis
Cell-type heterogeneity was corrected using the Reference-Free Adjustment for Cell-Type composition (ReFACTor) approach with the R software described by Rahmani and colleagues [54]. The association between blood methylation levels at cg05575921 and living in PM2.5 areas was determined using multiple linear regression analysis. Adjustments were made for sex, age, alcohol drinking, exercise, BMI, exposure to SHS, and cell type composition [54]. The inclusion of sex, age, alcohol drinking, exercise, and BMI as confounders is because they are common factors that have been associated with health. Moreover, DNA methylation predictors have been shown to correlate with these factors [55]. For instance, in previous studies, older age was significantly associated with lower cg05575921 methylation [24] and increased PM2.5-related mortality [56]. The male sex was significantly associated with lower cg05575921 methylation [24] while the female sex was significantly associated with an increased risk of PM2.5-related mortality [56]. The regression analysis was performed with the SAS 9.3 software (SAS Institute, Cary, NC).
Abbreviations
- AHRR:
-
Aryl hydrocarbon receptor repressor
- CpG:
-
Cytosine-phosphate-guanine
- DNA:
-
Deoxyribonucleic acid
- MOST:
-
Ministry of Science and Technology
- n :
-
Sample size
- PAH:
-
Polycyclic aromatic hydrocarbon
- PM2.5 :
-
Particulate matter < 2.5 microns in aerodynamic diameter
- SD:
-
Standard deviation
- SHS:
-
Second-hand smoke
- β :
-
Regression coefficient
References
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23.
Jin Z, Liu Y. DNA methylation in human diseases. Genes Dis. 2018;5(1):1–8.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nature Rev Genet. 2012;13:484.
Bojesen SE, Timpson N, Relton C, Smith GD, Nordestgaard BG. AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax. 2017;72(7):646–53.
Reynolds LM, Wan M, Ding J, Taylor JR, Lohman K, Su D, Bennett BD, Porter DK, Gimple R, Pittman GS. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis clinical perspective. Circ Cardiovasc Genet. 2015;8:707–16.
Gao X, Zhang Y, Saum K-U, Schöttker B, Breitling LP, Brenner H. Tobacco smoking and smoking-related DNA methylation are associated with the development of frailty among older adults. Epigenetics. 2017;12(2):149–56.
Kobayashi N, Miyauchi N, Tatsuta N, Kitamura A, Okae H, Hiura H, Sato A, Utsunomiya T, Yaegashi N, Nakai K. Factors associated with aberrant imprint methylation and oligozoospermia. Sci Rep. 2017;7:42336.
Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the sister study. Environ Health Perspect. 2014;122:673.
Zhu X, Li J, Deng S, Yu K, Liu X, Deng Q, Sun H, Zhang X, He M, Guo H. Genome-wide analysis of DNA methylation and cigarette smoking in a Chinese population. Environ Health Perspect. 2016;124:966.
Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, Martínez A, Narayan G, Kirsch I, Franklin W. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118(2):640–50.
Vogel CF, Haarmann-Stemmann T. The aryl hydrocarbon receptor repressor–more than a simple feedback inhibitor of AhR signaling: clues for its role in inflammation and cancer. Curr Opin Toxicol. 2017;2:109–19.
Lee D-H, Hwang S-H, Lim MK, Oh J-K, Yun EH, Park EY. Performance of urine cotinine and hypomethylation of AHRR and F2RL3 as biomarkers for smoking exposure in a population-based cohort. PloS One. 2017;12:e0176783.
Fasanelli F, Baglietto L, Ponzi E, Guida F, Campanella G, Johansson M, Grankvist K, Johansson M, Assumma MB, Naccarati A. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun. 2015;6:10192.
Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618.
Philibert R, Hollenbeck N, Andersen E, Osborn T, Gerrard M, Gibbons FX, Wang K. A quantitative epigenetic approach for the assessment of cigarette consumption. Front Psychol. 2015;6:656.
Philibert RA, Beach SR, Brody GH. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics. 2012;7(11):1331–8.
Andrysík Z, Vondráček J, Marvanová S, Ciganek M, Neča J, Pěnčíková K, Mahadevan B, Topinka J, Baird WM, Kozubík A. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: the role of polycyclic aromatic hydrocarbons. Mutat Res-Fund Mol M. 2011;714:53–62.
Ma Y, Li MD. Establishment of a strong link between smoking and cancer pathogenesis through DNA methylation analysis. Sci Rep. 2017;7(1):1811.
van der Zee SC, Fischer PH, Hoek G. Air pollution in perspective: health risks of air pollution expressed in equivalent numbers of passively smoked cigarettes. Environ Res. 2016;148:475–83.
Hung L-J, Chan T-F, Wu C-H, Chiu H-F, Yang C-Y. Traffic air pollution and risk of death from ovarian cancer in Taiwan: fine particulate matter (PM2. 5) as a proxy marker. Toxicol Environ Health A. 2012;75:174–82.
van Voorhis M, Knopp S, Julliard W, Fechner JH, Zhang X, Schauer JJ, Mezrich JD. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PloS One. 2013;8(12):e82545.
Castañeda AR, Pinkerton KE, Bein KJ, Magaña-Méndez A, Yang HT, Ashwood P, Vogel CF. Ambient particulate matter activates the aryl hydrocarbon receptor in dendritic cells and enhances Th17 polarization. Toxicol Lett. 2018;292:85–96.
Panni T, Mehta AJ, Schwartz JD, Baccarelli AA, Just AC, Wolf K, Wahl S, Cyrys J, Kunze S, Strauch K. Genome-wide analysis of DNA methylation and fine particulate matter air pollution in three study populations: KORA F3, KORA F4, and the Normative Aging Study. Environ Health Perspect. 2016;124:983.
Reynolds LM, Magid HS, Chi GC, Lohman K, Barr RG, Kaufman JD, Hoeschele I, Blaha MJ, Navas-Acien A, Liu Y. Secondhand tobacco smoke exposure associations with DNA methylation of the aryl hydrocarbon receptor repressor. Nicotine Tob Res. 2016;19:442–51.
Martin EM, Fry RC. Environmental influences on the epigenome: exposure-associated DNA methylation in human populations. Annu Rev Public Health. 2018.
Yeh H-L, Hsu S-W, Chang Y-C, Chan T-C, Tsou H-C, Chang Y-C, Chiang P-H. Spatial analysis of ambient PM2. 5 exposure and bladder cancer mortality in Taiwan. Int J Environ Res Public Health. 2017;14:508.
Chiang P, Chen CW, Hsieh DP, Chan T-C, Chiang H-C, Wen C-P. Lung cancer risk in females due to exposures to PM2. 5 in Taiwan. Open Epidemiol J. 2014;7.
Lo W-C, Shie R-H, Chan C-C, Lin H-H. Burden of disease attributable to ambient fine particulate matter exposure in Taiwan. J Formos Med Assoc. 2017;116:32–40.
Chen M-L, Mao I-F, Lin I-K. The PM2. 5 and PM10 particles in urban areas of Taiwan. Sci Total Environ. 1999;226:227–35.
Plusquin M, Guida F, Polidoro S, Vermeulen R, Raaschou-Nielsen O, Campanella G, Hoek G, Kyrtopoulos SA, Georgiadis P, Naccarati A. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ Int. 2017;108:127–36.
Abraham E, Rousseaux S, Agier L, Giorgis-Allemand L, Tost J, Galineau J, Hulin A, Siroux V, Vaiman D, Charles M-A. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ Int. 2018;118:334–47.
Bauer M. Cell-type-specific disturbance of DNA methylation pattern: a chance to get more benefit from and to minimize cohorts for epigenome-wide association studies. Int J Epidemiol. 2018.
Bauer M, Fink B, Thürmann L, Eszlinger M, Herberth G, Lehmann I. Tobacco smoking differently influences cell types of the innate and adaptive immune system—indications from CpG site methylation. Clin Epigenetics. 2016;8:83.
Philibert R, Hollenbeck N, Andersen E, McElroy S, Wilson S, Vercande K, Beach SR, Osborn T, Gerrard M, Gibbons FX. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front Psychol. 2016;7:55.
Philibert RA, Beach SR, Lei M-K, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5(1):19.
Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, Belvisi MG, Brown R, Vineis P, Flanagan JM. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Gen. 2012;22(5):843–51.
Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15(1):151.
Li S, Wong EM, Bui M, Nguyen TL, Joo J-HE, Stone J, Dite GS, Giles GG, Saffery R, Southey MC. Causal effect of smoking on DNA methylation in peripheral blood: a twin and family study. Clin Epigenetics. 2018;10(1):18.
Gao X, Jia M, Zhang Y, Breitling LP, Brenner H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7(1):113.
Chio C-P, Liao C-M, Tsai Y-I, Cheng M-T, Chou W-C. Health risk assessment for residents exposed to atmospheric diesel exhaust particles in southern region of Taiwan. Atmos Environ. 2014;85:64–72.
Fang G-C, Chang K-F, Lu C, Bai H. Estimation of PAHs dry deposition and BaP toxic equivalency factors (TEFs) study at Urban, Industry Park and rural sampling sites in central Taiwan, Taichung. Chemosphere. 2004;55(6):787–96.
Chen S-J, Liao S-H, Jian W-J, Lin C-C. Particle size distribution of aerosol carbons in ambient air. Environ Int. 1997;23(4):475–88.
Cho C-C, Hsieh W-Y, Tsai C-H, Chen C-Y, Chang H-F, Lin C-S. In vitro and in vivo experimental studies of PM2. 5 on disease progression. Int J Environ Res Public Health. 2018;15(7):1380.
Cavanagh J-AE, Trought K, Brown L, Duggan S. Exploratory investigation of the chemical characteristics and relative toxicity of ambient air particulates from two New Zealand cities. Sci Total Environ. 2009;407(18):5007–18.
Murillo-Tovar MA, Barradas-Gimate A, Arias-Montoya MI, Saldarriaga-Noreña HA. Polycyclic aromatic hydrocarbons (PAHs) associated with PM2.5 in Guadalajara, Mexico: environmental levels, health risks and possible sources. Environments. 2018;5(5):62.
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8(3):389–99.
Shahadin MS, Ab Mutalib NS, Latif MT, Catherine GM, Tidi H. Challenges and future direction of molecular research in air pollution-related lung cancers. Lung Cancer. 2018;118:69–75.
Fan C-T, Lin J-C, Lee C-H. Taiwan Biobank: a project aiming to aid Taiwan’s transition into a biomedical island. Pharmacogenomics. 2008;9(2):235–46.
Purpose Of Taiwan Biobank [https://www.twbiobank.org.tw/new_web_en/index.php]. Accessed 21 Mar 2018.
Bojovic B, Blancher C. Epigenetic analysis on Illumina EPIC arrays. Epigenetics. 2017;28:03.
Gujar H, Liang JW, Wong NC, Mozhui K. Profiling DNA methylation differences between inbred mouse strains on the Illumina Human Infinium MethylationEPIC microarray. PloS One. 2018;13(3):e0193496.
Infinium Assay Lab Setup and Procedures Guide [https://support.illumina.com/downloads/infinium-assay-lab-setup-and-procedures-11322460.html]. Accessed 23 Apr 2018.
Module, GenomeStudio Methylation v1. 8 User Guide [https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/genomestudio/genomestudio-2011-1/genomestudio-methylation-v1-8-user-guide-11319130-b.pdf. Accessed 27 Dec 2018.
Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, Oh S, Burchard EG, Eskin E, Zou J. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13(5):443.
McCartney DL, Hillary RF, Stevenson AJ, Ritchie SJ, Walker RM, Zhang Q, Morris SW, Bermingham ML, Campbell A, Murray AD. Epigenetic prediction of complex traits and death. Genome Biol. 2018;19(1):136.
Di Q, Dai L, Wang Y, Zanobetti A, Choirat C, Schwartz JD, Dominici F. Association of short-term exposure to air pollution with mortality in older adults. JAMA. 2017;318(24):2446–56.
Acknowledgements
Not applicable.
Funding
This work was supported by the Ministry of Science and Technology (MOST), Taiwan (MOST 106-EPA-F-016-001).
Availability of data and materials
The data that support the findings of this study are available from Taiwan Biobank but restrictions apply to the availability of these data, which were used under license for the current study, and so are not publicly available. Data are however available from the authors upon reasonable request and with permission of Taiwan Biobank.
Author information
Authors and Affiliations
Contributions
DMT and YPL conceived and designed the study. DMT, KJL, ONN, LYC, CCH, WMF, YCL, and TD performed the literature search. YPL, KJL, CL PHC, HWC SYH, WHL, WYK, and CCL acquired data and performed the data analysis. DMT, KJL, ONN, LYC, SYH, WHL, CCH, CCL, WMF, YCL, TD, and YPL interpreted the data. DMT wrote the manuscript. DMT, ONN, and YPL made critical revisions of the manuscript for important intellectual contents. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Chung Shan Medical University Institutional Review Board (CS2-17070).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Spearman correlation between PM2.5 concentrations (μg/m3) and mean methylation levels (beta values) in the northern, north-central, central, and southern areas. The methylation beta values decrease as PM2.5 levels increase. (DOCX 40 kb)
Additional file 2:
AHRR CpG sites significantly associated with PM2.5. (DOCX 20 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tantoh, D.M., Lee, KJ., Nfor, O.N. et al. Methylation at cg05575921 of a smoking-related gene (AHRR) in non-smoking Taiwanese adults residing in areas with different PM2.5 concentrations. Clin Epigenet 11, 69 (2019). https://doi.org/10.1186/s13148-019-0662-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-019-0662-9