
Abstract
The commensal, symbiotic, and pathogenic microbial community which resides inside our body and on our skin (the human microbiome) can perturb host energy metabolism and immunity, and thus significantly influence development of a variety of human diseases. Therefore, the field has attracted unprecedented attention in the last decade. Although a large amount of data has been generated, there are still many unanswered questions and no universal agreements on how microbiome affects human health have been agreed upon. Consequently, this review was written to provide an updated overview of the rapidly expanding field, with a focus on revealing knowledge gaps and research opportunities. Specifically, the review covered animal physiology, optimal microbiome standard, health intervention by manipulating microbiome, knowledge base building by text mining, microbiota community structure and its implications in human diseases and health monitoring by analyzing microbiome in the blood. The review should enhance interest in conducting novel microbiota investigations that will further improve health and therapy.
Similar content being viewed by others


What is microbiome?
Microorganisms often live in the form of a community. Furthermore, they can live in close association with complex organisms, such as plants and humans, by establishing commensal, ammensal, mutualistic, parasitic and/or pathogenic relationships with their hosts. The collection of such microorganisms is called microbiome or microbiota. Microflora has also been used but flora represents the kingdom Plantae therefore it is a misnomer.
In the original version, microbiome referred to the collection of microbes and their genomic contents. Microbiota indicated the microbial community in their host. But “microbiome” has frequently been used interchangeably with microbiota [1]. In this review, we focused mainly on bacterial microbiome with reference to either the collection of bacteria or their genomes, unless otherwise specified.
Microbiome can be found throughout the human body, ranging from the skin to the gut, and to previously considered as sterile environments such as the blood in circulation [2]. Various reports indicated that over 10,000 microbial species have been shown to occupy various parts of the human body [3, 4]. While diversity of microbes in the skin and vaginal sites are relatively low, great diversity can be found in other sites, e.g. the gut [6]. Consequently, impact of microbiome in human diseases and vice versa can be extensive. For example, chronic lung diseases can alter composition of lung microbiome which can subsequently influence host defense and immunity, thus leading to further exacerbation of the diseases [5]. Infection status has also been found to influence microbiome in the blood or the lung [6,7,8,9].
What is the gut microbiome?
The gut microbiome is the genetic material of all the microbes, e.g. bacteria, fungi, protozoa and viruses which live on and inside the digestive tracts of humans and other animals, including insects. In this review, we focused on the human gut microbiome and on bacterial composition.
The human gut microbiome has co-evolved with its host for millennia and, therefore, has been extensively involved with a variety of essential activities in the host, e.g. digestion and nutrition [10, 11], detoxification and body defense [12], maturation of the host immune system [11] and disease mediation [13,14,15,16,17]. Consequently, a large number of microbes with high diversity can be found in the mammalian gut, with most of them being Firmicutes and Bacteroidetes [18]. Such observation has been confirmed in different populations: Europeans and Americans [19], Koreans [20], Africans [21, 22], Danish but not Chinese [23]. The diversity can have specific implications for disease in different populations. For example, European and Chinese citizens with type 2 diabetes had different gut microbiome compositions [24], with the Chinese having more diverse species [24]. However, the reason of the major difference between the two populations, e.g. as related to age, environmental and genetic factors needs further investigation [25].
With diverse microorganisms, the gut microbiome contains millions of different genes [19]. Some of them may be acquired from environmental bacteria [10], indicating their metabolic diversity and versatility. Accordingly, three major genera have been reported as enterotypes: Bacteroides, Prevotella and Ruminococcus in the human gut as observed from 22 Europeans, 13 Japanese and 4 Americans [26]. Interestingly, similar bacterial ecosystems were also identified in mice and chimpanzees [27,28,29]. Their content in the human gut has been reported to be mainly influenced by their evolving change in the host and much less by age, gender, body weight, or race [26, 30]. However, a recent study reported that diet had more influence on metabolome than microbiome. In another context, some studies reported that Ruminococcus was a major ecotype [30,31,32], including one which analyzed data from native populations from different countries [33]. In particular, Enterobacteriaceae belonged to the third major ecotype among Taiwanese [34]. However, these discrepancies need to be clarified with more attention to sample size, and sampling methods and variations.
There are two major categories of microbes in the gut microbiota: (1) autochthonous microbes that seem to reside on the epithelium of colonic mucosa, and (2) allochthonous microbes that transiently pass the lumen as part of the digesta [35]. The functional roles of these “residents” and “passengers” are believed to be very different. Indeed, the ratio of autochthonous to non-autochthonous microbes has been proven useful to assess cirrhosis progression [36].
In general, host diet and phylogeny contribute to modifying the composition of gut microbial community in mammals and other species [18, 37, 38]. Indeed, genome-scale metabolic modeling show that variations in the diet of the host significantly modified the composition of the three representative human gut bacteria (B. thetaiotaomicron, E. rectale and M. smithii) [39]. For example, alcohol is a common dietary modulator of intestinal microbiota, as shown in experimental animals and humans [40,41,42,43,44]. In return, different composition of the three representative human gut bacteria influenced host metabolism and related diseases.
There are many reports which indicate that host genetics played an important role in determining the composition of microbiome [15, 45,46,47,48,49,50,51,52,53,54,55,56,57]. For example, several susceptibility loci were shared by inflammatory bowel disease [16, 17, 52], with infectious mycobacterial and staphylococcal organisms. These associations were validated from studies using the Gene Co-expression Network Analysis [58]. Therefore, investigations on the relationships among susceptibility, microbiome composition and disease development can provide valuable evidence to develop disease prevention protocols.
In the gut, a typical microbial product is lipopolysaccharide (LPS) which are produced by Gram-negative bacteria [6, 59,60,61,62] and are transported with chylomicrons [63]. LPS has been shown to be strong stimulators of innate immunity in organisms from lower- to higher-order animals [64]. For peritoneal dialysis patients, LPS level is used as an important indicator for survival. Indeed, a retrospective study of 300 patients show that plasma bacterial DNA levels were positively correlated with serum C-reactive proteins and LPS levels, and negatively correlated with survival rates [7]. These results indicate that both plasma LPS and bacterial DNA levels can be used as indicators for systemic inflammation and for prognosis. Another important microbial product is Trimethylamine (TMA). The oxidation product of TMA by hepatic flavine monoxygenases, trimethylamine N-oxide (TMAO), has influence on morbidity of patients [65]. These observations indicate that localized microbiome can cause far-reaching consequences.
Knowledge gaps and opportunities
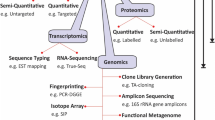
As mentioned earlier, stimulating observations in the new field of microbiome research has raised world-wide interest in the topic as well as many unanswered questions. Based on our review of the literature, we have identified several important issues and questions that may be useful for enhancement of novel research activities (Fig. 1).

Summary of knowledge gaps and opportunities in current study
Microbiota and animal physiology
There have been suggestions to treat the gut microbiome as our second genome, even as one of our tissues or organs. If the latter would be the case, the parenchyma (main tissue) and stroma (“sporadic” tissues) would have to be defined. Furthermore, it is necessary to find out how the microbiota are connected with each other. Should they be grouped with our biological system such as the immune, digestive, nervous or endocrine system? Can we gain insights into the adaptation of microbiota to their host by comparing different microbiota in the tree of life to better understand their inter-relationships?
Reports have shown that several physiological functions were protected directly by specific microbes via their control of epithelial cell proliferation and differentiation, and via their production of essential mucosal nutrients [66]. In addition, microbiota can protect physiological functions indirectly. For example, a certain gut bacteria caused behavioral abnormality by host metabolome [67]. Furthermore, the main fermentation products of gut microbiome which are short-chain fatty acids (SCFA) acetate, propionate and butyrate plus gases provided external resources for metabolic activities [68]. In addition to microbiota itself, host parameters such as lifestyle, diet, drug usage, genetics and immune activities could influence the composition as well as different consequences of host physiology [69]. Consequently, there should be additional risk factors which can influence association between host physiology and microbiota.
Optimal microbiome standard
Recently, a question was raised on reproducibility of investigations on gut microbiome research in experimental animals [70]. Some of the discrepancies can be due to biases on genetic and environmental factors [71]. For example, the lack of standardization in fecal microbiota transplantation protocol for multiple recurrent Clostridium difficile infection was a cause for reduced efficacy [72]. These concerns emphasize that environmental factors, rodent husbandry and treatment protocols must be standardized and be reproducible.
There have been recommendations to define microbiome composition inside an individual as a bacterial ecosystems [26], or “biomarkers” [33]. However, there are extensive variations from one individual to another. One example is the ratio of Firmicutes to Bacteroidetes (F:B) which was also affected by age [21, 73]. High F:B ratio has been reported to be associated with various pathological states [4, 11, 24, 74,75,76]. Therefore, it will be intriguing to investigate what would be the consequences if the F:B ratio is altered by adopting a vegetarian diet [77]. It should also be noted that liver and inflammatory bowel diseases can be associated with reduction in Firimutes but also with increase in Bacteroidetes [40, 78, 79]. In this context, what is the “normal” range of F:B ratio in a population and with respect to age? Consequently, what would be the standard composition of pathobiome [80] for healthy individuals?
Health intervention by manipulating microbiota
In recent years, the identification of prebiotics (a non-digestible food ingredient that promotes growth of beneficial microorganisms in the intestine), probiotics (a microorganism introduced into the body for its beneficial qualities) and synbiotics (a mixture of prebiotic and probiotic which selectively promotes growth) has aroused strong research and commercial interests. However, no studies have been conducted to address clinical beneficiary from probiotics intervention [81]. Consequently, a main focus for probiotics research is to validate the benefit and the mechanisms for physiological effects via clinical trials.
With regards to probiotics, Lactobacillus and Bifidobacterium are most commonly used for investigations [82]. Lactobacillus has been considered an option for preventing antibiotic-associated diarrhea in children [83]. For example, Lactobacillus casei were reported to inhibit growth of Helicobacter pylori [84]. In addition, co-colonization of Lactobacillus rhamnosus GG and Bifidobacterium lactis Bb12 promoted innate immune responses to human rotavirus [84]. Other Lactobacillus strains were used as potential treatment options for non-alcoholic fatty liver disease [84], type 2 diabetes [84], and urinary tract [84] and HIV infections [85, 86]. Although Lactobacillus species have been used in dairy food production safely for a long time [87], species resolution sometimes matter because certain Lactobacillus strains are tolerant in a low pH circumstance [82, 88], and others are associated with diseases [24, 74, 75, 89,90,91,92,93,94,95]. Therefore, it is crucial to investigate what specific genes or factors can make the difference among these members in the Lactobacillus bacteria.
There are at least 9 Bifidobacterium species that are commonly identified in the human gut [63]. In pathological conditions such as colorectal cancer, inflammatory bowel disease, irritable bowel syndrome and obesity, the relative abundance of Bifidobacterium species either changed significantly or, as a whole, decreased substantially when compared with other gut microbiota [96,97,98,99]. However, Bifidobacterium species are also widely recognized for their beneficial effects. Like Lactobacilus, Bifidobacteria is also a popular probiotic. For example, different Bifidobacteria have been used as therapy to relieve symptoms in some respiratory diseases, such as asthma in infants with atopic dermatitis [100] and cedar pollinosis [101, 102]. Bifidobacteria can also interact with intestinal cells by regulating immunity and inflammatory gene expression. B. longum was reported to regulate TNF-α and IL-1α expression in ulcerative colitis patients [103]. Oral administration of Bifidobacterium also improved tumor-specific immunity [104, 105]. Therefore, further investigations on Bifidobacterium, Lactobacillus and other microbes as probiotics and standardization of their usage can bring major benefits to individuals and to the healthcare system. Further inquiries may include: are these two species the only members in the “probiome”, and which microbiota would bring beneficial health effects to the host?
The application of probiotics can be beyond conventional arena for harm reduction. For example, microbial transplantation was used to restore healthy gut microbiome and to improve therapeutic efficacy for recurrent Clostridium difficile colitis [106]. In another study, a single commensal microbe, segmented filamentous bacterium protected mice from pathogenic effects of Citrobacter rodentium [107]. Alcohol consumption is another example. While laws and regulations are commonly used to limit alcohol consumption, it is more difficult to control consumption than to provide supplement. In this case, alcoholic liver disease intervention was successful from the administration of prebiotics, probiotics or synbiotics that modulated the composition of healthy and pathogenic intestinal microbiota [41, 108, 109]. Therefore, more investigations can be focused onto interventions using prebiotics, probiotics, synbiotics or microbiome transplantation, and onto their cost-effectiveness.
Knowledge base building by text mining
From our literature review, we recognized that several databases (e.g. Human Microbiome Project [HMP], The Integrative Human Microbiome Project [iHMP,], MetaHIT, Canadian Human Microbiome Initiative, and Australian Jumpstart Human Microbiome etc.) have been established. However, these databases are insufficient to archive the vast amount of published data. For example, there were no centralized resources which catalogued factors which influenced the gut microbiome composition. Having such a resource can be useful for conducting comprehensive and systematic data mining for host genetic, diet, disease, alcohol use, or other factors which can stimulate development of novel gut microbiome research. Moreover, statistical analyses, such as meta- or enrichment analyses can be made possible. To do this systematically, text mining is an option. Title and abstract data can be acquired in batches by software from the PubMed open access database. Sentence tokenization, and entity recognition for genes, microbiome, diet and environmental factors can also be conducted. Relationships among these factors (e.g. positive or negative effects) can be constructed and standard measures such as precision and recall can be used for evaluating text mining algorithms, with the accuracy as defined by two independent assessors. Recently, a free information system named @Minter can be used for analysis of abstracts and for inference microbial interactions base on Support Vector Machines with text-mining algorithm [110]. We expect that more and more platform or software can be used to concatenate different databases and to perform analysis with text-mining algorithm for increased efficiency of gut microbiome studies.
Microbiota community structure and its implications in human diseases
Microbes have been shown to interact extensively with each other within the human bodies [50, 111, 112]. For example, the Human Microbiome Project (HMP) cohort study reported competition between Porphyromonaceae and Streptococcus species in dental plaques, and between Prevotellaceae and Bacteroides in the guts; but possible complementation between Treponema and Prevotella in dental plaques [112]. In addition, co-existence of Candida fungi and H. pylori in the gastric mucosa was critical to the development of non-ulcer dyspepsia, gastric ulcer and duodenal ulcer [113].
Interactions can also be beneficial to the host. Through the production of polysaccharide A (PSA), Bacteroides fragilis protected its host from the induction of colitis via Helicobacter hepaticus infection [114]. Indeed, PSA has recently been shown to activate intestinal sensory neurons and thereby modulated peristalsis [115]. Administration of E. coli O21:H+ also protected mice from muscle wasting which was induced by infections [52].
Although it has been well-acknowledged that interactions between host and microbiome can significantly influence health and modulate clinical outcomes, more detailed mechanistic investigations are needed to better understand the important interactions and the opportunity for interventions. Investigations into interactions can borrow concepts from a seminal study in human disease network [116]. The then-hypothesis was if each human disorder had a distinct profile of microbiome, or ‘the pathobiome’ [80], the human disease network would be subdivided into many single nodes which corresponded to specific disorders or to small clusters of a few closely related disorders. Likewise, if microbial genes which were linked by disorder association with encoded proteins that interacted in functionally distinguishable modules, then the proteins within such disease modules would more likely interact with one another than with other proteins. Consequently, there would be significantly more protein–protein interactions, elevated gene ontology homogeneity and co-expression levels. Analyses of microbial co-existing relationships in the human and environment microbiomes [111, 112, 117] or spatial neighborhood [118] will provide useful reference resources to establish statistical significance of any novel pairs of bacterial groups. Drug disease network [119] can also be re-evaluated to offer new options for novel development of therapeutics.
Health monitoring by analyzing microbiome in the blood
Blood plasma has routinely been used to identify microbiome with the collection of bacteria and its products (e.g. nucleotide), for assessment of health status. The general assumption has been that nucleic acids originated mostly from gut microbiome with shedding into the blood. [120]. However, the existence of live microbiota in the blood circulation of apparently normal people was quite unexpected. For example, non-human small RNAs from Proteobacteria and fungus Hypocreales were detected in human blood samples [121]. An assay that detects and sequences plasma cell-free DNA (cfDNA) has been used to simultaneously monitor for infection and rejection in lung transplant recipients [122]. While the level of donor-derived cfDNA was strongly correlated with rejection, the level of cytomegalovirus-derived sequences cfDNA was indicative of infection. The small-molecular products from the gut microbiome can permeate the human serum and influence the rest of the human body [120]. Therefore, there are opportunities to investigate the role of blood microbiome in the disease process and the role of metabolome.
Some functional significance of microbiome in the blood circulation have recently been revealed [120]. For example, microvesicles which are laden with exogenous microbial RNA have the potential to function as signaling molecules in human plasma [123]. Non-human small RNAs from Proteobacteria and fungus Hypocreales were also detected in human blood samples [121]. Monitoring of nonhuman cell free DNA revealed undiagnosed infection which complicated prognosis [8]. Since alcohol has been found to increase intestinal permeability and it is a risk factor for endotoxemia, exploring the role of non-coding microbial RNAs in alcoholic liver disease is a natural direction for investigations, after adjusting for host genetic and other factors in alcohol elimination [124]. Cirrhosis dysbiosis ratio which is derived from stool microbiome can actually be tested to determine whether results obtained from blood microbiome are comparable to those from stool. For example, among 286 chronically HIV-infected individuals and AIDS patients, the LPS and 16S rDNA levels, and the percentage of CD8+, CD38+ and HLA-DR+ T cells were significantly higher than that from the uninfected controls [6]. Along with our previous reports [8, 9], health/disease status monitoring by assessing blood microbiome can be highly rewarding.
Conclusions
In this review, we have provided an overview of gut microbiome and some relevant research questions. Certainly, the use of molecular genetics, high-throughput procedures, bioinformatics and modeling can complement conventional techniques and fill existing knowledge gaps. How to make good use of the knowledge to benefit patient care and to optimize treatment plans (e.g. personalized) are valuable topics for investigation. Interventions such as long-term diet modification on controlling health risks [30] are appealing and cost- effective approaches. As long as sequencing cost can be further reduced, regular monitoring of personalized genome, transcriptome and microbiome will become more applicable in the near future. Collaborations among universities, hospitals and biotechnology companies can significantly enhance achieving these goals.
Change history
21 November 2019
Following publication of the original article in Gut Pathogens [1], it was brought to our attention that Dr. Ross Ka Kit Leung’s affiliation was incomplete. Dr. Leung is appointed as an Adjunct Professor of Guangzhou Medical University from January 1st 2017 to December 31st 2019. He would like to correct here the affiliation and acknowledge his role at the Guangzhou Medical University when this article was published.
References
Ursell LK, Metcalf JL, Parfrey LW, Knight R. Defining the human microbiome. Nutr Rev. 2012;70(Suppl 1):S38–44.
Proal AD, Albert PJ, Marshall TG. Inflammatory disease and the human microbiome. Discov Med. 2014;17(95):257–65.
Blaser MJ. Who are we? Indigenous microbes and the ecology of human diseases. EMBO Rep. 2006;7(10):956–60.
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3.
O’Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. 2016;196(12):4839–47.
Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, Landay A, Martin J, Sinclair E, Asher AI, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–85.
Kwan BC, Chow KM, Leung CB, Law MC, Cheng PM, Yu V, Li PK, Szeto CC. Circulating bacterial-derived DNA fragments as a marker of systemic inflammation in peritoneal dialysis. Nephrol Dial Transplant. 2013;28(8):2139–45.
Leung RK, Wu YK. Circulating microbial RNA and health. Sci Rep. 2015;5:16814.
Li SK, Leung RK, Guo HX, Wei JF, Wang JH, Kwong KT, Lee SS, Zhang C, Tsui SK. Detection and identification of plasma bacterial and viral elements in HIV/AIDS patients in comparison to healthy adults. Clin Microbiol Infect. 2012;18(11):1126–33.
Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464(7290):908–12.
Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587(Pt 17):4153–8.
Relman DA. The human microbiome: ecosystem resilience and health. Nutr Rev. 2012;70(Suppl 1):S2–9.
Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection. Science. 2015;347(6219):266–9.
Geddes K, Rubino SJ, Magalhaes JG, Streutker C, Le Bourhis L, Cho JH, Robertson SJ, Kim CJ, Kaul R, Philpott DJ, et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med. 2011;17(7):837–44.
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, Sigl V, Hanada T, Hanada R, Lipinski S, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487(7408):477–81.
Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482(7384):179–85.
Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, Golovkina TV. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334(6053):245–9.
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, et al. Evolution of mammals and their gut microbes. Science. 2008;320(5883):1647–51.
Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65.
Nam YD, Jung MJ, Roh SW, Kim MS, Bae JW. Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS ONE. 2011;6(7):e22109.
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107(33):14691–6.
Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, Turroni S, Biagi E, Peano C, Severgnini M, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun. 2014;5:3654.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–41.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60.
Tai N, Wong FS, Wen L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord. 2015;16(1):55–65.
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–80.
Hildebrand F, Nguyen TL, Brinkman B, Yunta RG, Cauwe B, Vandenabeele P, Liston A, Raes J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013;14(1):R4.
Moeller AH, Degnan PH, Pusey AE, Wilson ML, Hahn BH, Ochman H. Chimpanzees and humans harbour compositionally similar gut enterotypes. Nat Commun. 2012;3:1179.
Wang J, Linnenbrink M, Kunzel S, Fernandes R, Nadeau MJ, Rosenstiel P, Baines JF. Dietary history contributes to enterotype-like clustering and functional metagenomic content in the intestinal microbiome of wild mice. Proc Natl Acad Sci USA. 2014;111(26):E2703–10.
Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8.
Huse SM, Ye Y, Zhou Y, Fodor AA. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS ONE. 2012;7(6):e34242.
Zupancic ML, Cantarel BL, Liu Z, Drabek EF, Ryan KA, Cirimotich S, Jones C, Knight R, Walters WA, Knights D, et al. Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS ONE. 2012;7(8):e43052.
Gorvitovskaia A, Holmes SP, Huse SM. Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4:15.
Liang C, Tseng HC, Chen HM, Wang WC, Chiu CM, Chang JY, Lu KY, Weng SL, Chang TH, Chang CH, et al. Diversity and enterotype in gut bacterial community of adults in Taiwan. BMC Genom. 2017;18(Suppl 1):932.
Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146(6):1513–24.
Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940–7.
Miyake S, Ngugi DK, Stingl U. Diet strongly influences the gut microbiota of surgeonfishes. Mol Ecol. 2015;24(3):656–72.
Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127(2):423–33.
Shoaie S, Karlsson F, Mardinoglu A, Nookaew I, Bordel S, Nielsen J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep. 2013;3:2532.
Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54(2):562–72.
Mutlu E, Keshavarzian A, Engen P, Forsyth CB, Sikaroodi M, Gillevet P. Intestinal dysbiosis: a possible mechanism of alcohol-induced endotoxemia and alcoholic steatohepatitis in rats. Alcohol Clin Exp Res. 2009;33(10):1836–46.
Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK, Keshavarzian A. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. 2012;302(9):G966–78.
Queipo-Ortuno MI, Boto-Ordonez M, Murri M, Gomez-Zumaquero JM, Clemente-Postigo M, Estruch R, Cardona Diaz F, Andres-Lacueva C, Tinahones FJ. Influence of red wine polyphenols and ethanol on the gut microbiota ecology and biochemical biomarkers. Am J Clin Nutr. 2012;95(6):1323–34.
Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53(1):96–105.
Benson AK. Host genetic architecture and the landscape of microbiome composition: humans weigh in. Genome Biol. 2015;16:203.
Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci USA. 2010;107(44):18933–8.
Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, Vila AV, Deelen P, Vatanen T, Schirmer M, Smeekens SP, et al. The effect of host genetics on the gut microbiome. Nat Genet. 2016;48(11):1407–12.
Brinkman BM, Hildebrand F, Kubica M, Goosens D, Del Favero J, Declercq W, Raes J, Vandenabeele P. Caspase deficiency alters the murine gut microbiome. Cell Death Dis. 2011;2:e220.
Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45.
Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–99.
Igartua C, Davenport ER, Gilad Y, Nicolae DL, Pinto J, Ober C. Host genetic variation in mucosal immunity pathways influences the upper airway microbiome. Microbiome. 2017;5(1):16.
Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommeren S, Imhann F, Stempak JM, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014;6(12):107.
Kubinak JL, Stephens WZ, Soto R, Petersen C, Chiaro T, Gogokhia L, Bell R, Ajami NJ, Petrosino JF, Morrison L, et al. MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat Commun. 2015;6:8642.
Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Dore J, Raedler A, et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology. 2011;141(1):227–36.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6.
McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, Andreux PA, Bastiaansen JW, Wang X, Kachman SD, Auwerx J, et al. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLoS ONE. 2012;7(6):e39191.
Snijders AM, Langley SA, Kim YM, Brislawn CJ, Noecker C, Zink EM, Fansler SJ, Casey CP, Miller DR, Huang Y, et al. Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat Microbiol. 2016;2:16221.
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–24.
Balagopal A, Philp FH, Astemborski J, Block TM, Mehta A, Long R, Kirk GD, Mehta SH, Cox AL, Thomas DL, et al. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology. 2008;135(1):226–33.
Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–71.
Hofer U, Schlaepfer E, Baenziger S, Nischang M, Regenass S, Schwendener R, Kempf W, Nadal D, Speck RF. Inadequate clearance of translocated bacterial products in HIV-infected humanized mice. PLoS Pathog. 2010;6(4):e1000867.
Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev. 2013;26(1):2–18.
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50(1):90–7.
Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7(3):167–202.
Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120(7):1183–96.
Tappenden KA, Deutsch AS. The physiological relevance of the intestinal microbiota–contributions to human health. J Am Coll Nutr. 2007;26(6):679S–83S.
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451–63.
Flint HJ, Duncan SH, Scott KP, Louis P. Links between diet, gut microbiota composition and gut metabolism. Proc Nutr Soc. 2015;74(1):13–22.
Sommer F, Backhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–38.
Omary MB, Cohen DE, El-Omar EM, Jalan R, Low MJ, Nathanson MH, Peek RM Jr, Turner JR. Not all mice are the same: standardization of animal research data presentation. Cell Mol Gastroenterol Hepatol. 2016;2(4):391–3.
Hufeldt MR, Nielsen DS, Vogensen FK, Midtvedt T, Hansen AK. Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comp Med. 2010;60(5):336–47.
Seekatz AM, Aas J, Gessert CE, Rubin TA, Saman DM, Bakken JS, Young VB. Recovery of the gut microbiome following fecal microbiota transplantation. MBio. 2014;5(3):e00893-00814.
Mariat D, Firmesse O, Levenez F, Guimaraes V, Sokol H, Dore J, Corthier G, Furet JP. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123.
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103.
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE. 2010;5(2):e9085.
Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102(31):11070–5.
Matijasic BB, Obermajer T, Lipoglavsek L, Grabnar I, Avgustin G, Rogelj I. Association of dietary type with fecal microbiota in vegetarians and omnivores in Slovenia. Eur J Nutr. 2014;53(4):1051–64.
Farrell RJ, LaMont JT. Microbial factors in inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31(1):41–62.
Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut. 2004;53(1):1–4.
Vayssier-Taussat M, Albina E, Citti C, Cosson JF, Jacques MA, Lebrun MH, Le Loir Y, Ogliastro M, Petit MA, Roumagnac P, et al. Shifting the paradigm from pathogens to pathobiome: new concepts in the light of meta-omics. Front Cell Infect Microbiol. 2014;4:29.
Sanders ME, Guarner F, Guerrant R, Holt PR, Quigley EM, Sartor RB, Sherman PM, Mayer EA. An update on the use and investigation of probiotics in health and disease. Gut. 2013;62(5):787–96.
Fontana L, Bermudez-Brito M, Plaza-Diaz J, Munoz-Quezada S, Gil A. Sources, isolation, characterisation and evaluation of probiotics. Br J Nutr. 2013;109(Suppl 2):S35–50.
Goldenberg JZ, Lytvyn L, Steurich J, Parkin P, Mahant S, Johnston BC. Probiotics for the prevention of pediatric antibiotic-associated diarrhea. Cochrane Database Syst Rev. 2015;12:CD004827.
Sgouras D, Maragkoudakis P, Petraki K, Martinez-Gonzalez B, Eriotou E, Michopoulos S, Kalantzopoulos G, Tsakalidou E, Mentis A. In vitro and in vivo inhibition of Helicobacter pylori by Lactobacillus casei strain Shirota. Appl Environ Microbiol. 2004;70(1):518–26.
Anukam KC, Osazuwa EO, Osadolor HB, Bruce AW, Reid G. Yogurt containing probiotic Lactobacillus rhamnosus GR-1 and L. reuteri RC-14 helps resolve moderate diarrhea and increases CD4 count in HIV/AIDS patients. J Clin Gastroenterol. 2008;42(3):239–43.
Hummelen R, Changalucha J, Butamanya NL, Koyama TE, Cook A, Habbema JD, Reid G. Effect of 25 weeks probiotic supplementation on immune function of HIV patients. Gut Microbes. 2011;2(2):80–5.
Bernardeau M, Guguen M, Vernoux JP. Beneficial lactobacilli in food and feed: long-term use, biodiversity and proposals for specific and realistic safety assessments. FEMS Microbiol Rev. 2006;30(4):487–513.
Bosch M, Rodriguez M, Garcia F, Fernandez E, Fuentes MC, Cune J. Probiotic properties of Lactobacillus plantarum CECT 7315 and CECT 7316 isolated from faeces of healthy children. Lett Appl Microbiol. 2012;54(3):240–6.
Gan XT, Ettinger G, Huang CX, Burton JP, Haist JV, Rajapurohitam V, Sidaway JE, Martin G, Gloor GB, Swann JR, et al. Probiotic administration attenuates myocardial hypertrophy and heart failure after myocardial infarction in the rat. Circ Heart Fail. 2014;7(3):491–9.
Gomez-Guzman M, Toral M, Romero M, Jimenez R, Galindo P, Sanchez M, Zarzuelo MJ, Olivares M, Galvez J, Duarte J. Antihypertensive effects of probiotics Lactobacillus strains in spontaneously hypertensive rats. Mol Nutr Food Res. 2015;59(11):2326–36.
Karlsson FH, Fak F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Backhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245.
Kawase M, Hashimoto H, Hosoda M, Morita H, Hosono A. Effect of administration of fermented milk containing whey protein concentrate to rats and healthy men on serum lipids and blood pressure. J Dairy Sci. 2000;83(2):255–63.
Simon MC, Strassburger K, Nowotny B, Kolb H, Nowotny P, Burkart V, Zivehe F, Hwang JH, Stehle P, Pacini G, et al. Intake of Lactobacillus reuteri improves incretin and insulin secretion in glucose-tolerant humans: a proof of concept. Diabetes Care. 2015;38(10):1827–34.
Tanida M, Yamano T, Maeda K, Okumura N, Fukushima Y, Nagai K. Effects of intraduodenal injection of Lactobacillus johnsonii La1 on renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Neurosci Lett. 2005;389(2):109–14.
Yamashiro K, Tanaka R, Urabe T, Ueno Y, Yamashiro Y, Nomoto K, Takahashi T, Tsuji H, Asahara T, Hattori N. Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS ONE. 2017;12(2):e0171521.
Collado MC, Isolauri E, Laitinen K, Salminen S. Effect of mother’s weight on infant’s microbiota acquisition, composition, and activity during early infancy: a prospective follow-up study initiated in early pregnancy. Am J Clin Nutr. 2010;92(5):1023–30.
Gueimonde M, Ouwehand A, Huhtinen H, Salminen E, Salminen S. Qualitative and quantitative analyses of the bifidobacterial microbiota in the colonic mucosa of patients with colorectal cancer, diverticulitis and inflammatory bowel disease. World J Gastroenterol. 2007;13(29):3985–9.
Kerckhoffs APM, Samsom M, van der Rest ME, de Vogel J, Knol J, Ben-Amor K, Akkermans LMA. Lower Bifidobacteria counts in both duodenal mucosa-associated and fecal microbiota in irritable bowel syndrome patients. World J Gastroentero. 2009;15(23):2887–92.
Mylonaki M, Rayment NB, Rampton DS, Hudspith BN, Brostoff J. Molecular characterization of rectal mucosa-associated bacterial flora in inflammatory bowel disease. Inflamm Bowel Dis. 2005;11(5):481–7.
van der Aa LB, van Aalderen WM, Heymans HS, Henk Sillevis Smitt J, Nauta AJ, Knippels LM, Ben Amor K, Sprikkelman AB, Synbad Study G. Synbiotics prevent asthma-like symptoms in infants with atopic dermatitis. Allergy. 2011;66(2):170–7.
Odamaki T, Xiao JZ, Iwabuchi N, Sakamoto M, Takahashi N, Kondo S, Miyaji K, Iwatsuki K, Togashi H, Enomoto T, et al. Influence of Bifidobacterium longum BB536 intake on faecal microbiota in individuals with Japanese cedar pollinosis during the pollen season. J Med Microbiol. 2007;56(Pt 10):1301–8.
Xiao JZ, Kondo S, Yanagisawa N, Takahashi N, Odamaki T, Iwabuchi N, Iwatsuki K, Kokubo S, Togashi H, Enomoto K, et al. Effect of probiotic Bifidobacterium longum BB536 [corrected] in relieving clinical symptoms and modulating plasma cytokine levels of Japanese cedar pollinosis during the pollen season. A randomized double-blind, placebo-controlled trial. J Investig Allergol Clin Immunol. 2006;16(2):86–93.
Furrie E, Macfarlane S, Kennedy A, Cummings JH, Walsh SV, O’Neil DA, Macfarlane GT. Synbiotic therapy (Bifidobacterium longum/Synergy 1) initiates resolution of inflammation in patients with active ulcerative colitis: a randomised controlled pilot trial. Gut. 2005;54(2):242–9.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–9.
Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–84.
Bakken JS, Borody T, Brandt LJ, Brill JV, Demarco DC, Franzos MA, Kelly C, Khoruts A, Louie T, Martinelli LP, et al. Treating Clostridium difficile infection with fecal microbiota transplantation. Clin Gastroenterol Hepatol. 2011;9(12):1044–9.
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98.
Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM. Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology. 2004;39(5):1441–9.
Bull-Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, Gobejishvili L, Joshi-Barve S, Ayvaz T, Petrosino J, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS ONE. 2013;8(1):e53028.
Lim KM, Li C, Chng KR, Nagarajan N. @MInter: automated text-mining of microbial interactions. Bioinformatics. 2016;32(19):2981–7.
Chaffron S, Rehrauer H, Pernthaler J, von Mering C. A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 2010;20(7):947–59.
Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8(7):e1002606.
Karczewska E, Wojtas I, Sito E, Trojanowska D, Budak A, Zwolinska-Wcislo M, Wilk A. Assessment of co-existence of Helicobacter pylori and Candida fungi in diseases of the upper gastrointestinal tract. J Physiol Pharmacol. 2009;60:33–9.
Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–5.
Mao YK, Kasper DL, Wang B, Forsythe P, Bienenstock J, Kunze WA. Bacteroides fragilis polysaccharide A is necessary and sufficient for acute activation of intestinal sensory neurons. Nat Commun. 2013;4:1465.
Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci USA. 2007;104(21):8685–90.
Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK, Keshavarzian A. Colonic microbiome is altered in alcoholism. Am J Physiol Gastr L. 2012;302(9):G966–78.
Earle KA, Billings G, Sigal M, Lichtman JS, Hansson GC, Elias JE, Amieva MR, Huang KC, Sonnenburg JL. Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe. 2015;18(4):478–88.
Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25(10):1119–26.
Potgieter M, Bester J, Kell DB, Pretorius E. The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol Rev. 2015;39(4):567–91.
Beatty M, Guduric-Fuchs J, Brown E, Bridgett S, Chakravarthy U, Hogg RE, Simpson DA. Small RNAs from plants, bacteria and fungi within the order Hypocreales are ubiquitous in human plasma. BMC Genom. 2014;15:933.
De Vlaminck I, Martin L, Kertesz M, Patel K, Kowarsky M, Strehl C, Cohen G, Luikart H, Neff NF, Okamoto J, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci USA. 2015;112(43):13336–41.
Wang K, Li H, Yuan Y, Etheridge A, Zhou Y, Huang D, Wilmes P, Galas D. The complex exogenous RNA spectra in human plasma: an interface with human gut biota? PLoS ONE. 2012;7(12):e51009.
Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16(4):667–85.
Authors’ contributions
DL and RKKL designed the study, reviewed relevant articles and wrote the manuscript. DL and RKKL made the figure. WG and WWA participated in the design of the study and helped draft and revise the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thanked Qi Zhang for her literature search for the very first draft of this manuscript.
Competing interests
The authors declare that they have no conflict of interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This project was supported by the Fund for Less Developed Regions of the National Natural Science Foundation of China (Grant No. 81660012) and the National Key Research and Development Program of China (Grant No. 2017YFC1309302).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liang, D., Leung, R.KK., Guan, W. et al. Involvement of gut microbiome in human health and disease: brief overview, knowledge gaps and research opportunities. Gut Pathog 10, 3 (2018). https://doi.org/10.1186/s13099-018-0230-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-018-0230-4