
Abstract
Heterocyclic 1,3-diazine nucleus is a valuable pharmacophore in the field of medicinal chemistry and exhibit a wide spectrum of biological activities. PharmMapper, a robust online tool used for establishing the target proteins based on reverse pharmacophore mapping. PharmMapper study is carried out to explore the pharmacological activity of 1,3-diazine derivatives using reverse docking program. PharmMapper, an open web server was used to recognize for all the feasible target proteins for the developed compounds through reverse pharmacophore mapping. The results were analyzed via molecular docking with maestro v11.5 (Schrodinger 2018-1) using GTPase HRas as possible target. The molecular docking studies displayed the binding behavior of 1,3-diazine within GTP binding pocket. From the docking study compounds s3 and s14 showed better docked score with anticancer potency against cancer cell line (HCT116). Hence, the GTPase HRas may be the possible target of 1,3-diazine derivatives for their anticancer activity where the retrieved information may be quite useful for developing rational drug designing. Furthermore the selected 1,3-diazine compounds were evaluated for their in vitro anticancer activity against murine macrophages cell line. 1,3-Diazine compounds exhibited good selectivity of the compounds towards the human colorectal carcinoma cell line instead of the murine macrophages. The toxicity study of the most active compounds was also performed on non cancerous HEK-293 cell line.

Similar content being viewed by others


Introduction
Heterocyclic compounds play the vital role in pharmaceutical field due to their specific chemical reactivity and block the normal functioning of biological receptors. A large number of 1,3-diazine derivatives are reported to exhibit various biological activities i.e. anticancer [1], antibacterial [2], anti-inflammatory, analgesic [3], antimicrobial activity [4]. 1,3-Diazine nucleus is the building unit in DNA and RNA thus 1,3-diazine based compounds exhibit diverse biological activities. Thus 1,3-diazine and its derivative attract the researchers to further explore their biological activities [5].
According to World Health Organization (WHO) reports, cancer is one of the leading causes of death worldwide and is projected to continuously rising, with approximately 11.5 million deaths in 2030. The main types of cancer are of body organs like lung, stomach, colorectal, liver and breast. Cancer treatment includes psychosocial support, surgery, radiotherapy, chemotherapy that is aimed at curbing the disease as well as improving the quality of patient’s life [6]. Malignancy arises due to transformation of the genetic material of a normal cell, followed by successive mutations, ultimately leading to the uncontrolled division of cells. Drug resistance is a phenomenon that results when diseases become tolerant to pharmaceutical treatments. Drug resistance occurs through various mechanisms like drug inactivation, drug target alteration, drug efflux, DNA damage repair, cell death inhibition [7].
In modern drug discovery, molecular docking is now a day’s an advanced computational technique used to study the ligand–receptor interactions using docking software and uses conformational and electrostatic interactions to measures it. Molecular docking programs perform a search algorithm in which the conformation of the ligand is evaluated until the convergence to the minimum energy is reached. With the various docking strategies, the ligand specificity against a particular target (receptor) can be calculated computationally in which best fitting ligand can be used for further lead optimization process. The docking score (affinity scoring function), ΔG [U total in kcal/mol], is the sum of the electrostatic and van der Waals energies to rank the candidate poses to determine their binding potentialities. Docking score is calculated terms of negative energy [8, 9]. The heterocyclic pyrimidine derivatives displayed good anticancer potency against HCT116 cancer cell line [10,11,12].
Based on the above mentioned facts, in the present study, the reverse docking program was used to recognize the drug target for the anticancer activity of 1,3-diazine derivatives (identified in an earlier study [13]) using PharmMapper web server application tool. The receptor (target), GTPase HRas was found with good fitness score against cancer. The validation of the indicated target was done with molecular docking using maestro v11.5.
Experimental
Data set
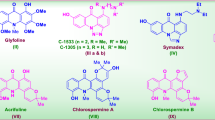
The data set of reported 1,3-diazine derivatives (s1–s16) have good anticancer activity against human colorectal carcinoma cancer cell line (HCT116) were selected from the earlier study for the establishment of the pharmacophore model. The synthesis of the reported compounds is shown in synthetic Scheme 1. The physicochemical properties and structural elucidations are shown in Tables 1 and 2, respectively. The molecular structures of the selected data set of 1,3-diazine derivatives and their anticancer screening results are shown in Table 3 [13].

Synthetic scheme for the synthesized 1,3-diazine derivatives (s1–s16)
Ligand preparation
Ligand preparation is done using the maestro v11.5 LigPrep module to deliver the best results, the docked structures must be good representations of the actual ligand structures as they would appear in a complex of protein–ligand. This implies that the structure must fulfill the following requirements for Glide docking program. They have to be 3-dimensional (3D). Glide only modifies the ligand’s internal torsional coordinates during docking, so the remaining geometric parameters need to be optimized in advance. They must each consist of a single molecule without covalent receptor bonds, with no accompanying fragments, such as counter ions and solvent molecules. They have to be filled with all their hydrogen (valences). For physiological pH values (around 7), they must have a suitable protonation state [14, 15].
Protein preparation
The protein chosen for the molecular docking study of selected data set of 1,3-diazine derivatives, GTPase HRas (PDB Id: 2CL7) was obtained from the Protein Data Bank (Additional file 1). The typical structure file imported from the PDB is not suitable for immediate use in performing calculations for molecular modeling. A typical PDB structure file consists of heavy atoms and may include a co-crystallized ligand, molecules of water, ions of metal and cofactors. In the protein preparation wizard, protein was prepared where protein was preprocessed, optimized and minimized. The outcome is refined, hydrogenated ligand and ligand–receptor complicated structures that are appropriate for use with other Schrödinger modules [16].
Grid generation
Maestro v11.5 receptor grid generation module (Schrodinger 2018-1) is used to generate grid. A grid is generated around the binding site already occupied by the co-crystallized ligand so that it is feasible to exclude co-crystallized ligand and to attach new molecule to the same binding site to study the docking of 1,3-diazine derivatives [17].
Docking study
After generating the glide grid zip file and preparing the ligands, docking was performed in the maestro v11.5 glide module. The series of ligands (1,3-diazines) was tested using additional accuracy (XP) via GTP binding site. XP Module conducts more accurate molecular docking of the selected molecules of 1,3-diazine nucleus. The size of the dataset is reduced as the precision of the docking increase at each stage. In the maestro v11.5, the XP parameters such as docking score glide energy and glide emodel value were calculated [18, 19].
Anticancer evaluation
Anticancer activity of the synthesized compounds was evaluated on the cell line of murine macrophage (RAW 264.7) by SRB assay (Table 3) [20]. The murine macrophage cell line was seeded at 7000 cells/well on the 96 flat bottom well plate and allowed to be activated overnight. Then the cells were exposed for 72 h to the respective compounds and subjected to the SRB test. Then treated cells were located in trichloroacetic acid and stained in SRB dye [0.4% (w/v) mixed with 1% acetic acid]. The plate’s optical density was read with a microplate reader at 570 nm.
Cell toxicity evaluation
The cell toxicity study of the selected compounds was performed on non-cancer cell line, i.e. human embryonic kidney (HEK 293). In Dulbecco’s modified Eagle medium (10% heat inactivated FBS) human embryonic kidney cells were maintained. Penicillin and streptomycin antibiotics were added and placed in a 5% CO2 incubator for colorimetric-based using MTT assay at 37 °C. Compounds s3, s9, s13–s16 were seeded on a 24-h 96-well plate with five thousand HEK-293 cells (viability 98%). Wells have been added to MTT 5 mg/ml for 4 h after 24 h incubation [21]. Using the Synergy/HTX MultiScan reader (BioTek) absorbance at 580 nm was registered and the lethal dose LD50 was calculated and the selectivity index (SI) calculated.
Results and discussion
Target recognition
An open web portal, PharmMapper was used through reverse pharmacophore mapping to account for all possible compound targets [22]. PharmMapper sets the feasible potential targets based on the reverse pharmacophore mapping of given 1,3-diazine compounds. It compares the pharmacophores of the compounds given to the BindingDB, TargetBank, DrugBank, PDTD with 16,159 druggable and 51,431 ligandable pharmacophore models in built pharmacophore models database of 23,236 proteins. It offers outcomes in the form of Z score based on the resemblance of pharmacophore of specified compounds with the recognized target pharmacophore model as well as the significance of target protein in illnesses and signs are provided as well [23, 24]. In order to define its potential drug target, the most active compounds s3 and s14 were presented to PharmMapper. Depending on their role in cancer initiation and progression, target protein was chosen.
Target identification
From the information collection, the PharmMapper (http://59.78.96.61/pharmmapper) received compounds s3 and s14 showing the advanced anti-carcinogenic activity. The pharmacophores of the potent compounds s3 and s14 were compared with the built-in pharmacophore model database. PharmMapper compared the pharmacophores of the potent compounds s3 and s14 with the created-in pharmacophore model database and generated 250 protein target information with their fitness score and pharmacophoric characteristics, indication and importance of each protein. 250 Protein retrieved were ranked based on their fitness score. Top five proteins with fitness score more than 5.0 were studied to establish the possible target protein for compounds s3 and s14 and target selection was done based upon the role of protein in cancer disease (Table 4). GTPase HRas protein with 15 pharmacophoric characteristics (8 acceptor, 5 donor and 2 negative) scored 5.424 out of the top five proteins, was discovered to play a crucial part in cancer determinism. Another protein with a healthy fitness value, but as shown in Table 4, did not account for any disease. The GTPase HRas protein function is governed by the GTP where GTP becomes GDP. GTP-based HRas protein’s mechanism of action is discovered to function by signal transduction in regulating cell division and cell growth. Mutation in HRas has been shown to lead to different cancer types such as bladder, Costello syndrome, bladder cancer, etc. Because HRas belongs to the oncogene family, healthy cells can become cancerous [25]. GTPase HRas has been further assessed through the docking program for the binding affinity for the studied 1,3-diazine derivatives.
Docking
Previously, GTPase HRas and 1,3-diazine derivatives were ready for docking and then docked using maestro v11.5 Glide module (Schrodinger 2018-1). GTP was maintained as docking control with docked score = − 10.434 and glide energy = − 80.151 in order to score the compounds to be studied. The docking is performed using PDB Id: 2CL7 (Fig. 1) in the same binding region of already co-crystallized GTP ligand. All 1,3-diazine compounds were scored using flexible docking (XP docking) where compounds used GTP as docking control. Minimization of docked compounds was carried out within the binding site and the most stable orientation was analyzed with the lowest possible energy. Docking score of the compounds is shown in Table 5. The results of PharmMapper and molecular docking showed the specificity of 1,3-diazine compounds for the protein GTPase HRas. Compounds demonstrated excellent interaction with GTPase HRas and binding affinity. If we look at the binding mode of most active compounds s3 and s14 within the binding region, compound s3 has docked in the binding pocket score (− 2.14) and glide energy (− 56.46) and hydrogen bond formation with crucial amino acid residue Gly60 with oxygen atom; Compound s14 has docked score (− 1.603) and glide force (− 66.638) and hydrogen bond formation in the binding pocket with vital amino acid residue Gly60 (Table 6, Figs. 2 and 3). Thus the docking results suggested that the compounds of 1,3-diazine could be of great interest in successful chemotherapy. The GTPase HRas may therefore be the possible target for their anticancer potential of 1,3-diazine derivatives. The experimental research will be carried out to validate the affinity to target protein and the binding mode of inhibition of compounds. The docking results of the data set and GTPas shown in Additional files 2, 3.

Pictorial presentation 3D (a) and ligand interaction diagram 2D (b) of GTP

Pictorial presentation 3D (a) and ligand interaction diagram 2D (b) of compound s3

Pictorial presentation 3D (a) and ligand interaction diagram 2D (b) of compound s14
Anticancer screening results
Table 3 shows the comparison between HCT116 and RAW 264.7 of the IC50 values of the 1,3-diazine derivatives (s1–s16). 1,3-Diazine compounds showed good selectivity of compounds to the human colorectal cell line of carcinoma instead of the murine macrophages. The compounds of IC50 1,3-diazine versus RAW 264.7 were all beyond the largest concentration tested. Among the compounds tested (on the RAW 264.7 line of murine macrophages), compounds s11 and s16 showed better potency against the cell line of murine macrophages. The control drug had an antiproliferative impact on both lines of the cell.
Cell toxicity analysis
These were screened against ordinary human embryonic renal cell line (HEK-293) for the selectivity index calculation of the chosen compounds. Compounds have been dissolved into DMSO solution of 0.1%. The concentration of the compounds (2 µM, 4 µM, 6 µM, 8 µM and 10 µM) was diluted. The cells were incubated with these compounds for 24 h and at IC50 for growth inhibition of each researched compound, nearly 100% of HEK-293 cells were feasible. Results showed the important difference in viability after 24 h with (P < 0.01) between the treated test compound and the control cells (at zero concentration). The 50% of neurons were feasible at the chosen compounds lethal dose (LD50) of 8.55 to 8.18 µM. As we understand, the LD50 value greater than the IC50 will be the selectivity that meant that the compounds could have better safety for each of the six compounds as the IC50 is much smaller than the LD50 compounds. Each compound’s selectivity index suggested better safety for each compound (Table 7).
Conclusion
Computational methods such as PharmMapper and molecular docking are cost-effective and time-saving instrument used respectively to determine target protein and generate docking data. GTPase HRas was discovered to be a target receptor among the top five scored protein to study the antiproliferative potential of more effective compounds s3 and s14. In the GTPase HRas protein binding site, the further docking of 1,3-diazine compounds produced the docking poses of the most active compound and GTP used as positive control. In addition, compounds s11 and s16 showed stronger anti-cancer activity against the cell line of murine macrophages. The impact of most active compounds on the cell viability of non-cancerous HEK-293 cells has also been investigated in the current research. The findings showed a stronger selectivity index at the corresponding concentration of IC50 against the HEK 293 cell lines. Study proposed that after experimental assessment, the compound may be safer as an anticancer. 1,3-Diazine compounds (s1–s16) showed excellent selectivity of the compounds towards the cell line of human colorectal carcinoma instead of the murine macrophages. After further experimental validation, most active compounds may be safer to use. The research suggested that GTPase HRas protein with a stronger selectivity index could be the possible target protein of 1,3-diazine compounds.
Abbreviations
- HCT116:
-
human colorectal carcinoma116
- RAW 264.7:
-
murine macrophaga 264.7
- PDB:
-
Protein Data Bank
- GTP\HR-protein:
-
guanosine-5′-triphosphate/Harvey rat-protein
- XP:
-
extra precision
- 2D:
-
2 dimensional
- 3D:
-
3 dimensional
- SRB:
-
sulforhodamine B
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HEK-293:
-
human embryonic kidney-293
- SI:
-
selectivity index
- LD50 :
-
lethal dose50
- 5-Fu:
-
5-fluorouracil
- RNA:
-
ribonucleic acid
- DNA:
-
deoxyribonucleic acid
- NMR:
-
nuclear magnetic resonance
- IR:
-
infrared
- MS:
-
mass spectrum
- CHN:
-
carbon hydrogen nitrogen
- Str:
-
starching
References
Ghorab MM, Alsaid MS (2016) Anticancer activity of some novel thieno[2,3-d]pyrimidine derivatives. Biomed Res 27(1):110–115
Cieplik J, Stolarczyk M, Pluta J, Gubrynowicz O, Bryndal I, Lis T, Mikulewicz M (2011) Synthesis and antibacterial properties of pyrimidine derivatives. Acta Pol Pharm Drug Res 68(1):57–65
Nofal ZM, Fahmy HH, Zarea ES, El-Eraky W (2011) Synthesis of new pyrimidine derivatives with evaluation of their anti-inflammatory and analgesic activities. Acta Pol Pharm Drug Res 68(4):507–517
Gupta YK, Gupta V, Singh S (2013) Synthesis, characterization and antimicrobial activity of pyrimidine based derivatives. J Pharm Res 7:491–495
Bhat AR (2017) Biological activity of pyrimidine derivatives: a review. Organ Med Chem 2(2):1–4
Ott JJ, Ullrich A, Mascarenhas M, Stevens GA (2010) Global cancer incidence and mortality caused by behavior and infection. J Public Health 33(2):223–233
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014) Drug resistance in cancer: an overview. Cancers 6:1769–1792
Pagadala NS, Syed K, Tuszynski J (2017) Software for molecular docking: a review. Biophys Rev 9:91–102
Bassyouni F, Hefnawi ME, Rashed AE, Rehim MA (2017) Molecular modeling and biological activities of new potent antimicrobial, anti-inflammatory and anti-nociceptive of 5-nitro indoline-2-one derivatives. Drug Des 6(2):1–6
Yousif MNM, El-Sayed WA, Abbas HAS, Awad HM, Yousif NM (2017) Anticancer activity of new substituted pyrimidines, their thioglycosides and thiazolopyrimidine derivatives. J Appl Pharm Sci 7(11):021–032
Mohamed MS, Youns MM, Ahmed NM (2015) Evaluation of anticancer activity of some thiouracil derivatives. Int J Pharm Biol Sci 5(4):159
Mohamed AM, Al-Qalawi HRM, El-Sayed WA, Arafa WAA, Alhumaimess MS, Hassan AK (2015) Anticancer activity of newly synthesized triazolopyrimidine derivatives and their nucleoside analogs. Acta Pol Pharm 72(2):307–318
Kumar S, Lim SM, Ramasamy K, Mani V, Shah SAA, Narasimhan B (2018) Design, synthesis, antimicrobial and cytotoxicity study on human colorectal carcinoma cell line of new 4,4′-(1,4-phenylene)bis(pyrimidin-2-amine) derivatives. Chem Cent J 12(73):1–13
Kumar S, Singh J, Narasimhan B, Shah SAA, Lim SM, Ramasamy K, Mani V (2018) Reverse pharmacophore mapping and molecular docking studies for discovery of GTPase HRas as promising drug target for bis-pyrimidine derivatives. Chem Cent J 12(106):1–11
Driessche GVD, Fourches D (2017) Adverse drug reactions triggered by the common HLA-B*57:01 variant: a molecular docking study. J Cheminform 9(13):1–17
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols and influence on virtual screening enrichments. J Comput Aid Mol Des 27(3):221–234
Sharma V, Sharma PC, Kumar V (2016) In silico molecular docking analysis of natural pyridoacridines as anticancer agents. Adv Chem 2016:1–9
Friesner RA, Murph RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem 49:6177–6196
Lenselink EB, Louvel J, Forti AF, van Veldhoven JPD, de Vries H, Mulder-Krieger T, McRobb FM, Negri A, Goose J, Abel R, van Vlijmen HWT, Wang L, Harder E, Sherman W, IJzerman AP, Beuming T (2016) Predicting binding affinities for GPCR ligands using free-energy perturbation. ACS Omega 1:293–304
Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR (1990) New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82:1107–1112
Selvaraj V, Bodapati S, Murray E, Rice KM, Winston N, Shokuhfar T, Zhao Y, Blough E (2014) Cytotoxicity and genotoxicity caused by yttrium oxide nanoparticles in HEK293 cells. Int J Nanomed 9:1379–1391
Ge S-M, Zhan DL, Zhang S-H, Song L-Q, Han W-W (2016) Reverse screening approach to identify potential anti-cancer targets of dipyridamole. Am J Transl Res 8(12):5187–5198
Wang X, Shen Y, Wang S, Li S, Zhang W, Liu X, Lai L, Pei J, Li H (2017) PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res 45(W1):W356–W360
Wang X, Pan C, Gong J, Liu X, Li H (2016) Enhancing the enrichment of pharmacophore-based target prediction for the polypharmacological profiles of drugs. J Chem Inf Model 56(6):1175–1183
Colicelli J (2004) Human RAS superfamily proteins and related GTPases. Sci STKE 250:1–13
Acknowledgements
The authors are thankful to Head, Department of Pharmaceutical Sciences, Maharshi Dayanand University, Rohtak, for providing necessary facilities to carry out this research work.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
BN, SK and DS—performed computational docking study, SML, KR, VM and SAAS—performed cytotoxicity study of synthesized compounds. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1.
Web link for GTPase HRas protein.
Additional file 2.
Docking results of the data set.
Additional file 3.
Docking results, Pictorial presentation and Ligand interaction diagram of GTPas.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kumar, S., Sharma, D., Narasimhan, B. et al. Computational approaches: discovery of GTPase HRas as prospective drug target for 1,3-diazine scaffolds. BMC Chemistry 13, 96 (2019). https://doi.org/10.1186/s13065-019-0613-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13065-019-0613-8