
Abstract
Background
E-proteins encoded by Tcf3, Tcf4, and Tcf12 are class I basic helix-loop-helix (bHLH) transcription factors (TFs) that are thought to be widely expressed during development. However, their function in the developing brain, specifically in the telencephalon remains an active area of research. Our study examines for the first time if combined loss of two E-proteins (Tcf3 and Tcf12) influence distinct cell fates and oligodendrocyte development in the mouse telencephalon.
Methods
We generated Tcf3/12 double conditional knockouts (dcKOs) using Olig2Cre/+ or Olig1Cre/+ to overcome compensatory mechanisms between E-proteins and to understand the specific requirement for Tcf3 and Tcf12 in the ventral telencephalon and during oligodendrogenesis. We utilized a combination of in situ hybridization, immunohistochemistry, and immunofluorescence to address development of the telencephalon and oligodendrogenesis at embryonic and postnatal stages in Tcf3/12 dcKOs.
Results
We show that the E-proteins Tcf3 and Tcf12 are expressed in progenitors of the embryonic telencephalon and throughout the oligodendrocyte lineage in the postnatal brain. Tcf3/12 dcKOs showed transient defects in progenitor cells with an enlarged medial ganglionic eminence (MGE) region which correlated with reduced generation of embryonic oligodendrocyte progenitor cells (OPCs) and increased expression of MGE interneuron genes. Postnatal Tcf3/12 dcKOs showed a recovery of OPCs but displayed a sustained reduction in mature oligodendrocytes (OLs). Interestingly, Tcf4 remained expressed in the dcKOs suggesting that it cannot compensate for the loss of Tcf3 and Tcf12. Generation of Tcf3/12 dcKOs with Olig1Cre/+ avoided the MGE morphology defect caused by Olig2Cre/+ but dcKOs still exhibited reduced embryonic OPCs and subsequent reduction in postnatal OLs.
Conclusion
Our data reveal that Tcf3 and Tcf12 play a role in controlling OPC versus cortical interneuron cell fate decisions in MGE progenitors in addition to playing roles in the generation of embryonic OPCs and differentiation of postnatal OLs in the oligodendrocyte lineage.
Similar content being viewed by others


Introduction
Oligodendrogenesis is a critical event in brain development that results in the generation of oligodendrocyte progenitor cells (OPCs) that differentiate into oligodendrocytes (OLs) which produce myelin that is crucial for the health of the axons [1, 2]. During development, OL generation is under precise spatiotemporal regulation, determined by appropriate activation of signaling pathways and transcription factors (TFs) [3, 4]. Furthermore, single cell RNA sequencing studies have revealed a new level of differential gene expression as neural progenitors, OPCs, and OLs go through distinct stages during brain development [5,6,7]. Identifying key regulators at these distinct stages is an ongoing effort in the OL field.
The class II basic-helix-loop-helix (bHLH) TFs Ascl1 and Olig2 are regulators early in oligodendrogenesis as knockouts show robust defects in the embryonic generation of OPCs in brain and spinal cord [8,9,10,11,12,13,14,15]. In addition, stage specific conditional knockouts and in vitro cultures revealed that these genes as well as Olig1 play roles at later stages in postnatal OPC generation and/or OL differentiation [11, 16,17,18,19,20,21]. These class II bHLH TFs are known to heterodimerize with class I bHLH TFs, called E-proteins, to bind DNA [22,23,24]. Despite the numerous studies identifying roles for class II bHLH TFs during OL development, the class I bHLH TFs remain understudied in this cell lineage.
Even though E-proteins were thought to be widely expressed, Tcf4 has been characterized to show enriched regional expression in the developing cortex and hippocampus in the telencephalon [25, 26]. In addition, enriched Tcf4 and Tcf12 expression has also been identified in the medial ganglionic eminence (MGE) in the ventral most progenitors in the telencephalon [27, 28]. Previous in vitro studies have revealed that Tcf3 and Tcf12 exhibit cell type expression in cultured oligodendrocyte precursors [29]. Additional in vitro studies showed that co-expression of Tcf3 with either Ascl1, Olig1, or Olig2 can influence later stages of OL development [30]. Tcf12 was also identified as one of the TFs expressed in OPCs derived from human neural progenitor cells [31]. Tcf4 is the most studied E-protein in the developing brain [32]. Loss-of-function mutations of Tcf4 cause the neurodevelopmental disorder called Pitt Hopkins syndrome and Tcf4 KO mouse models have revealed roles for Tcf4 in the cortex and hippocampus [25, 33,34,35,36]. In addition, Tcf4 KOs showed defects in the OL lineage cells, specifically in later OL differentiation stages [37, 38]. Despite these studies on Tcf4, there continues to be a lack of understanding of the in vivo requirement for Tcf3 and Tcf12 in oligodendrogenesis. In fact, Tcf3 and Tcf12 have redundant and compensatory roles and single knockouts revealed no CNS defects [39,40,41,42,43].
We investigated the importance of Tcf3 and Tcf12 in OL development by creating a double conditional knockout (dcKO) mouse model using Olig2cre/+ mice. We found a simultaneous loss of Tcf3 and Tcf12 resulted in transient abnormal morphology of the MGE, a loss of OPCs, and an increase of MGE interneuron genes during embryogenesis. However, at postnatal stages, OPC numbers recovered from embryonic time points, but mature OL (mOL) populations remained decreased in the dcKOs. Later embryonic deletion of Tcf3 and Tcf12 using Olig1cre/+ avoided the MGE phenotype but the embryonic OPC reduction and postnatal OL differentiation defects remained. Our study concludes that Tcf3 and Tcf12 play roles in ventral telencephalon cell fate decisions and specifically in the OL lineage for embryonic OPC generation and the generation of postnatal OLs.
Materials and methods
Animals
Protocols for animal experiments using mice were approved by the Institutional Animal Care and Use Committee at the Cincinnati Children’s Hospital Medical Center and carried out in accordance with National Institutes of Health guidelines. Tcf12tm3Zhu ;Tcf3tm4Zhu (Stock #024511), referred to as Tcf3/12flox in this manuscript, Rosatom/+ (Stock #007914), and FoxG1IRES − Cre/+ (Stock #029690) mice were obtained from Jax and genotyped from published protocols on Jax website. Olig2cre/+ mice were previously described [44] and provided by Y. Yoshida. Olig1cre/+ mice were previously described [17]. For specific embryonic collections, vaginal plug indicates embryonic day 0.5 during timed matings. For BrdU treatment, BrdU was dissolved into PBS at a 10 mg/1mL dilution. Pregnant females were administered 0.1 mg of BrdU for every 1 g of weight through intraperitoneal injection, 30 min before embryonic dissection. Embryo and adult collections and processing for histology were completed as previously described [45,46,47].
Immunohistochemistry and immunofluorescence
Primary antibodies were used at the following concentrations: Animal-Antibody (Dilution, Company, catalog #), Rbt-Ascl1 (1:5,000 Abcam, #AB74065), Gt-βgal (1:1,000, Biogenesis, #2282), Rbt-βgal (1:1000, Biogenesis, #4600 − 1509), Rat-BrdU (1:250, Abcam, #AB6326), Rbt-CNPase (1:500, Cell Signaling, #5664), MS-CC1 (1:100, Calbiochem #OP80-100UG), GP-Gsx1 (1:5000, [48]), Rbt-Gsx2 (1:4000, [49]), Ch-MBP (1:500, Aves, #AvesMBP), Gt-mCherry (1:1000, Biorbyt, #ORB11618), MS-Neurofilament (1:100, DSHB, #2H3), Rbt-Nkx2.1 (TTF1) (1:2000, Seven Hills Bioreagents, # WRAB-1231), Rbt-Nkx2.2 (1:1000, Abcam, #AB191077), Rbt-Olig2 (1:1000, Millipore Sigma, #AB9610), Gt-Pdgfrα (1:500, R&D Systems, #AF1062), Gt-Sox10 (1:500, Santa Cruz, catalog #SC-17,342), Rbt-Tcf4 (1:500, ProteinTech, #22337-1-AP), Rbt-Tcf12 (1:1000, ProteinTech ,#14419-1-AP). The mouse-neurofilament 2H3 antibody was obtained from the Developmental Studies Hybridoma Bank (DSHB) at the University of Iowa.
Fluorescent stains were cover slipped with SouthernBiotech DAPI Fluoromount-G to stain for DAPI.
In situ hybridization was completed as described [50]. The plasmid to generate Plp1 anti-sense probe was previously described [11]. The following primers were used to generate in situ probes:
Lhx6: Fwd - ATGCACTTCTCACCAGAGGC, Rev w/ T3 sequence - ATTAACCCTCACTAAAGG GGAGACGTCTGACTGCAACA,
Tcf12: Fwd - TCTCGAATGGAAGACCGC, Rev w/ T3 sequence - ATTAACCCTCACTAAAGGCTCCCTCCTGCCAGGTTT
Tcf3: Fwd – CCCCAACTACGATGCAGG, Rev w/ T3 sequence - ATTAACCCTCACTAAAGGCCGGGCAGAGATATGGTG
Tcf4: Fwd - GCGGTCTACGCTCCTTCA, Rev w/ T3 sequence - ATTAACCCTCACTAAAGGTGGGTTCAAGTCAGGGGA
All bright field pictures were captured on a Leica DM2500 microscope with a DMC6200 camera with 1.0x c-mount or DFC500 camera with 0.7x c-mount using Leica Acquisition Software. Fluorescence images were captured on a Nikon C2 confocal microscope using Nikon Elements software. Representative images were selected from at least n = 3 animals for each genotype and each stain. “Controls” refer to wild type animals or animals with a mixed status of floxed Tcf3 and/or Tcf12 alleles with no Cre, unless otherwise noted.
Quantification
Immunofluorescent images for quantification in Fig. 2 were taken at 20x magnification with a 1.5x zoom. 4 serial sections of 3 different control animals were examined for each set of E-protein/OL marker stain. Images were taken at 20x for quantification for most stains, with the following exceptions: MBP/NFM (10x), Nkx2.1 (5x for MGE images, BrdU (40x), Tcf4/Olig2 (10x at E15, 20x with 1.5 zoom for P21), Tcf4/DAPI (20x with 1.5 zoom). High magnification images were taken in corpus callosum, unless otherwise specified. For every data set, at least 3 controls and 3 mutants were analyzed. 4 serial sections of the region were imaged for quantification. Raw images were processed through Cell Profiler to identify the number of cells in the image, identify the area of the image occupied by the stains, or stain intensity, as indicated by the graph. Graphs were generated in Prism 9.0 GraphPad.
Statistics
Differences in controls and conditional double knockouts were assessed using Student’s two-tailed unpaired t-test. Results were considered significant if p-value was less than 0.05. Mean results and p-value are indicated in Results section. Bar graphs represent mean and error bars represent SEM. P-values are also indicated within bar graphs with * = p < 0.05, ** = p < 0.01, *** = p < 0.001 and **** = p < 0.0001. Statistical analyses were performed using Prism 9.0 GraphPad.
Results
E-protein expression in telencephalon development and oligodendrogenesis
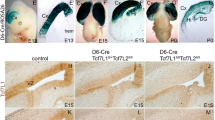
The genes encoding the three E-proteins were originally thought to be ubiquitously expressed [23, 24]. However, recent research suggests that some of these genes may show distinct regional enrichment during development of the telencephalon. For example, Tcf4 is robustly expressed in the developing cortex of the dorsal telencephalon, in the hippocampus of the medial telencephalon, and in the globus pallidus of the ventral telencephalon [25,26,27]. In addition, Tcf12 expression is enriched in the developing MGE of the ventral telencephalon [28, 51]. However, unlike Tcf4 and Tcf12, Tcf3 expression is not regionally enriched and is present in both dorsal and ventral progenitors of the telencephalon [52]. We utilized in situ hybridization for Tcf3, Tcf4, and Tcf12 at different developmental stages to directly compare E-protein gene expression during development of the telencephalon. The three E-protein genes have clear expression in the ventricular zone (VZ) throughout the telencephalon where the earliest neural progenitors reside at E13.5, E15.5, and E18.5 (Fig. 1A-I). In addition to the VZ expression, Tcf4 and Tcf12 are also enriched in the MGE at E13.5 (Fig. 1B, C) which is consistent with previous reports [27, 28]. At E15.5, Tcf3 and Tcf12 share similar expression patterns, exhibiting enrichment around the VZ (Fig. 1D,F). At this stage, Tcf4 begins to take on a distinct expression pattern, specifically enriched in the VZ, MGE, and in the maturing cortical regions (Fig. 1E; see white asterisks for cortex). Tcf3 and Tcf12 remain expressed in the VZ (E18.5) and forming subventricular zone (SVZ) at P7 (Fig. 1G, I, J, L, see black arrows for VZ/SVZ). Tcf4 expression shows the same VZ/SVZ enrichment at E18.5 and P7 as Tcf3 and Tcf12, as well as continued expression in the cortex (Fig. 1H, K, see black arrows for VZ/SVZ) consistent with previous reports [25, 26]. High magnification of the corpus callosum at P7 revealed expression of all three E-protein genes within the forming white matter track where oligodendrocytes are enriched (Fig. 1M-O), which is consistent with previous research [38]. Our comparative expression analysis in the developing telencephalon revealed that all three E-protein genes are widely expressed in the developing VZ/SVZ and in scattered cells of the forming white matter. Interestingly, Tcf4 and Tcf12 show regionally enriched expression in the MGE of the ventral telencephalon while Tcf4 was the only gene that showed robust regional enrichment in the maturing cortex of the dorsal telencephalon.

E-protein expression during telencephalon development. In situ hybridization for Tcf3 (A, D, G, J, M), Tcf4 (B, E, H, K, N), and Tcf12 (C, F, I, L, O). Representative images are shown at E13.5 (A-C), E15.5 (D-F), E18.5 (G-I), and P7 (J-O). At E13.5 and E15.5, all three E-proteins are enriched in the ventricular zone (VZ) in the CTX, LGE, and MGE (A-F). In addition, Tcf4 and Tcf12 also show high expression within the subventricular zone (SVZ) of the MGE at E13.5 and E15.5 (B-C, E-F). Enrichment for the E-proteins in the VZ continues at E18.5 and the SVZ at P7 (see black arrows G-L). Tcf4 specifically is highly enriched in the cortex at E15.5-P7 (see white asterisk, E, H, K). High magnification images of the corpus callosum at P7 reveal E-protein expression in the white matter track (M-O, black dashed box in J-L indicate the representative areas for M-O). Scale bar in F = 500 μm (for A-F), I = 600 μm (for G-I), L = 1 mm (for J-L), O = 200 μm (for M-O). LGE = lateral ganglionic eminence, MGE = medial ganglionic eminence, CTX = cortex, STM = striatum
Previous studies have analyzed E-protein gene expression in the oligodendrocyte lineage from RNA sequencing databases and protein expression from in vitro glial cultures [29, 38]. To better grasp the in vivo expression in the postnatal brain and oligodendrocytes, specifically in the scattered cells of the forming white matter, we examined immunofluorescence double stains of the E-proteins and markers of different OL stages. Due to a lack of suitable Tcf3 antibodies for immunofluorescence, a Tcf3 reporter was used where the Tcf3fx/+ allele has a lacZ reporter under control of the Tcf3 promoter that is out of frame until Cre recombination [53]. We utilized Tcf3fx/+; FoxG1IRES − Cre/+ mice to recombine lacZ into frame throughout the telencephalon under control of the Tcf3 promoter. Control mice were used to examine Tcf4 and Tcf12 expression. Both the Tcf3fx/+; FoxG1IRES − Cre/+ and control mice were analyzed at P21. Double staining with Olig2 and either β-gal (for Tcf3), Tcf4, or Tcf12 in the corpus callosum revealed that all three E-proteins are expressed within the OL lineage, specifically 56.8%±7.51% of Olig2 + cells express Tcf3, 74.9%±7.46% express Tcf4, and 59.4%±8.30% express Tcf12 (n = 3) (Fig. 2A-C). Using Pdgfrα as a marker of OPCs, we show that 20.4%±8.50% of Pdgfrα + cells express Tcf3, 55.9%±8.22% express Tcf4, and 29.0%±6.81% express Tcf12 (n = 3) (Fig. 2D-F). Nkx2.2 is a marker for OPCs transitioning into pre-myelinating immature oligodendrocytes (iOLs). We show that 52.5%±5.11% of Nkx2.2 + cells express Tcf3, 55.8%±5.76% express Tcf4 and 69.8%±6.80% express Tcf12 (n = 3) (Fig. 2G-I). Lastly, CC-1 is used as a marker for maturing iOLs and mOLs. We show that 38.4%±9.06% of CC-1 + cells express Tcf3, 46.4%±7.25% express Tcf4 and 54.8%±4.68% express Tcf12 (n = 3) (Fig. 2J-L). These data demonstrate that the E-proteins are expressed at all stages of oligodendrogenesis (Fig. 2M), which is in line with a previous in vitro analysis showing Tcf3 and Tcf12 are expressed in A2B5 + and O4 + rat oligodendrocyte precursors and more recently that Tcf4 is expressed in Pdgfrα+, O4+, and Mbp + cells from cultured rat oligodendroglial cells [29, 38]. In addition, our data is also in line with a searchable sc-RNA seq. gene expression data base at mature adult brain stages showing Tcf3, Tcf4, and Tcf12 expression during stages in the oligodendrocyte lineage [5].

E-protein expression throughout the oligodendrocyte lineage in the corpus callosum. Double immunofluorescence for Tcf4 (B, E, H, K), and Tcf12 (C, F, I, L) with OL lineage markers. β-gal was used as a reporter for Tcf3 in Tcf3fx/+;FoxG1IRES-Cre/+mice, where lacZ is recombined into frame under the Tcf3 promoter, and double stained for OL lineage markers (A, D, G, J). Olig2 was used to mark the entire OL lineage. All three E-proteins have some overlapping expression with Olig2 (see yellow arrows, A-C). Pdgfrα was used as a marker for OPC populations (see yellow arrows for overlap with E-proteins, D-F). Nkx2.2 marks maturing OPC populations (see yellow arrows for overlap, G-I). CC1 was used to define maturing OL populations (see yellow arrows for overlap, J-L). Quantification for percentage of OL lineage markers co-expressing the respective E-protein (M). Scale bar in L = 50 μm (for A-L)
Tcf3/12 dcKOs have deficits in OPC production
Recent studies have established that Tcf4 plays a key role in regulating appropriate numbers of oligodendrocyte lineage cells and is required for later stages of OL differentiation and myelination [37, 38]. No requirements for Tcf3 and Tcf12 have been identified in the OL lineage. In fact, previous analysis of Tcf3−/− and Tcf12−/− knockout mice and Tcf3/12+/− double heterozygous mice did not find any gross morphological differences in the mutant brains [43]. This is perhaps not surprising because Tcf3 and Tcf12 are well described in their ability to compensate for one another in a variety of tissue systems [39,40,41,42,43]. Tcf3−/−, Tcf12−/−, and Tcf3/12+/− mutations are lethal before breeding age likely from defects in the developing immune system or outside of the CNS, making the generation of a double Tcf3/12 knockout impossible [43, 54]. Therefore, to examine the cell type specific role for Tcf3 and Tcf12 in the developing telencephalon and during oligodendrogenesis without the complication of compensation, we generated a double conditional knockout (dcKO) of Tcf3/12 using Olig2Cre/+mice to target deletion in the ventral telencephalon where the majority of embryonic OPCs emerge and through the entire embryonic and postnatal OL lineage. We have previously used this approach to target the RASopathy gene Ptpn11 in the early oligodendrocyte lineage [47]. Analysis of Tcf3 gene expression and Tcf12 protein expression confirm robust recombination of Tcf3 and Tcf12 alleles in the ventral telencephalon of the dcKOs at E15.5 (Fig. 3A-D).

Tcf3/12 double conditional knock out using Olig2cre/+results in fewer oligodendrocyte progenitor cells. Confirmation of Tcf3/12 double conditional KO using Olig2Cre/+was validated by in situ hybridization with probe against Tcf3 (A-B) and immunostaining with an antibody against Tcf12 (C-D), showing loss of the respective E-protein gene expression in the double conditional knockouts (dcKOs). Olig2 marks neural progenitor cells in the VZ of the MGE and LGE as well as the OL lineage cells away from the VZ. At E15.5, Olig2 is increased in the VZ of the MGE and decreased in the parenchyma of dcKOs compared to controls (red asterisk shows the increased MGE region, E-F, K). Pdgfrα and Sox10 are both markers of OPCs and are largely absent in dcKOs (G-K). At E18.5, remnants of the increased MGE Olig2 expression remains visible, while Olig2 expression outside the VZ is decreased in the dcKOs compared to controls (black arrow for MGE remnant, L-M, R). Pdgfrα and Sox10 remain severely decreased in dcKOs compared to controls at E18.5 (N-R). Quantification of markers at E15.5 in the parenchyma (K) and E18.5 in the striatum (R). Scale bar in J = 500 μm (for A-J) and Q = 1 mm (for L-Q). LGE = lateral ganglionic eminence, MGE = medial ganglionic eminence, CTX = cortex, STM = striatum
To assess for the generation of embryonic OPCs from the ventral telencephalon, we stained for the OPC markers Olig2, Pdgfrα, and Sox10 [8, 9, 55,56,57,58,59]. Olig2 is first expressed in the VZ progenitors in the MGE and remains expressed in OPCs of the parenchyma [8, 9, 56]. Pdgfrα and Sox10 are expressed in OPCs and are both downstream of Olig2 [10, 58, 60, 61]. At E15.5, a dramatic reduction of OPC production is observed in the dcKOs compared to controls (Fig. 3E-K). During oligodendrogenesis, Olig2 + progenitors migrate out of the VZ in the ventral telencephalon and begin to express OPC markers Pdgfrα and Sox10 [12, 62]. The dcKOs express Olig2 in the VZ of the MGE, but show a 42.7% loss of Olig2 in the striatum away from the VZ (Fig. 3E-F,K, p = 0.0013). At E15.5, dcKOs show a severe depletion of OPCs marked by Pdgfrα and Sox10, with a 98.6% decrease of Pdgfrα (p < 0.0001) and 91.4% decrease of Sox10 (p < 0.0001) (Fig. 3G-K). This reduction of OPC numbers in the dcKOs continues at later embryonic stages right before birth. The number of Olig2 + cells in the striatum are reduced by 32.7% at E18.5 (Fig. 3L-M,R, p = 0.0005). However, Pdgfrα and Sox10 + cells remain severely depleted, with the number of Pdgfrα + cells reduced by 90.8% (Fig. 3N-O,R, p < 0.0001) and Sox10 expression reduced by 77.6% (Fig. 3P-R, p < 0.0001) in the dcKOs compared to controls. Unlike the robust defect in the Tcf3/12 dcKO, we did not observe OPC defects analyzing Olig2 + cells in the striatum of Tcf3 or Tcf12 single cKOs at E18.5 (Control = 106.7 ± 2.81 compared to Tcf3 cKO = 95.42 ± 4.01 and Tcf12 cKO = 94.75 ± 6.01 cells per field, p = 0.1350). This is in line with previous studies on germline single knockouts for Tcf3 or Tcf12 that did not find obvious CNS defects [43].
Interestingly, Olig2 expression in the VZ of the MGE revealed abnormal morphology in dcKOs at E15.5 (Fig. 3F, see red asterisks for the dorsal and ventral limits of the MGE), which is also apparent in the negative stained regions for Tcf3 and Tcf12 expression (Fig. 3B, D, see red asterisks for the dorsal and ventral limits of the MGE). This phenotype does persist to some extent at E18.5, where the morphological remnant of the expanded MGE remains apparent (see black arrow, Fig. 3M). Thus, dcKO embryos have a defect in OPC generation or maturation as they maintain expression of Olig2 in ventral progenitor areas and to some extent in the parenchyma cells but the OPC markers Pdgfrα and Sox10, which are expressed after Olig2, are severely reduced at all embryonic stages.
Postnatal Tcf3/12 dcKOs have reduced markers of mOL
Although OPC production is widespread throughout the telencephalon by late embryonic stages, OL differentiation and the expression of maturing myelin markers occurs largely after birth [63]. To determine if Tcf3/12 dcKOs exhibit OL differentiation defects, we examined the P7 stage as an early time point for gene expression of the myelin marker Plp1 to label differentiation and Nkx2.2 to label differentiating OPCs [64]. Plp1 expression can be seen in the corpus callosum and sparsely in the striatum and cortex in controls (Fig. 4A). Therefore, we focused on the corpus callosum/white matter to address the oligodendrocyte markers at this stage. The dcKOs have a severe deficit in Plp1 positive cells with an 85.0% reduction compared to controls in the corpus callosum and few positive cells were observed in the dcKO striatum or cortex (p < 0.0001, Fig. 4A-B, I). Nkx2.2 expression showed a 56.0% decrease in the number of positive cells in the dcKOs compared to controls (p < 0.0001, Fig. 4G-H, I). These data suggest that Tcf3/12 dcKOs exhibit a defect in the production of maturing OLs. Surprisingly, OPCs marked by Pdgfrα recovered to control levels in dcKOs by P7 in the corpus callosum (p = 0.7283, Fig. 4E-F, I). The same recovery was also observed in the cortex and striatum which contains more sparsely labeled cells (data not shown). Analysis of Olig2 to label the entire OL lineage (OPCs and OL) revealed only a 15.8% decrease in the dcKOs compared to control, likely reflecting the improvement in OPC numbers and the decrease of the mature cell types (p = 0.0277, Fig. 4C-D, I). However, it should be noted that Olig2 also labels a small population of astrocyte progenitors during the first week after birth [65]. Despite this, the Olig2 results are in line with the other oligodendrocyte lineage markers that show in a short time frame from E18.5 to P7, the OPC numbers in the dcKO recover to control levels but significant deficits appear in the differentiation of OPCs to OLs.

Tcf3/12 double conditional knock out mice show reduction of mature oligodendrocytes at postnatal time point P7. Tcf3/12 dcKOs have a dramatic reduction of mature oligodendrocytes, marked by Plp1 gene expression (A-B, I). Olig2, marking the entire OL lineage, is also decreased in the CC of dcKOs compared to controls (C-D, I). OPC marker, Pdgfrα, shows no difference in expression between controls and dcKOs, indicating a recovery from the E18.5 time point (E-F, I). Nkx2.2, expressed in differentiating OPCs, is also decreased at P7 in the dcKOs compared to controls (G-I). Images (C-H) are high magnification representatives of the corpus callosum in controls and dcKOs. Quantification of respective markers, counted in the corpus callosum (I). Scale bar in B = 1 mm (for A-B) and H = 100 μm (for C-H). CTX = Cortex, CC= Corpus Callosum, STM = Striatum
The postnatal recovery of OPCs in the dcKO suggests that the OL differentiation defect at P7 may be temporary and the timing of OL generation might be delayed. However, analysis of dcKOs at P21 near the end of developmental myelination revealed the Plp1 defect was maintained with a 32.5% decrease in dcKOs compared to controls in the corpus callosum (p < 0.0001, Fig. 5, A-B,O). At this stage, robust expression of myelin proteins, such as CNPase and MBP, can be observed as an additional method to analyze OL differentiation and the dcKOs show reduce staining density in the cortex (Fig. 5C-F). We detected a 31.8% reduction in MBP expression in the dcKOs compared to control cortical areas, while NFM expression to label the axons is unchanged (p = 0.0035, p = 0.82 respectively, Fig. 5E-F, O). Despite the deficit in both OL numbers and myelin gene and protein expression, dcKO animals did not exhibit any obvious hypomyelination phenotypes, like shivering or shaking. Similar to the P7 result, there is a decrease in Olig2 + cells labeling the entire OL lineage with a 38.5% reduction in cells at P21 (p-value = 0.0085, Fig. 5G-H,O). This decrease likely reflects the reduction in mature OL numbers since OPCs labeled with Pdgfrα were not changed in dcKOs compared to controls (p = 0.3430, Fig. 5I-J,O). In contrast to the phenotype at P7, the number of Nkx2.2 + cells is comparable between controls and dcKOs at P21 (p = 0.8402, Fig. 5K-L,O). This may indicate a continuing recovery of cells within the OL lineage. However, Nkx2.2 labels mature and differentiating OPCs and newly formed OLs which is a relatively small population at P21, so any reduction in this population might be masked by the existing mature OPCs. To determine if the reduction of Plp + cells at P7 and P21 observed in dcKOs is transient and a delay in differentiation, we analyzed 2 month old animals (P60). The reduction in Plp1 is maintained at these later stages with a 43.0% reduction in dcKOs compared to controls (p < 0.0001, Fig. 5M-N,O). This indicates that mOL recovery is not occurring and suggests Tcf3 and Tcf12 play a role in the appropriate generation of OLs at postnatal stages, in addition to a requirement for embryonic OPC generation at earlier stages.

Reduction of mature oligodendrocytes in Tcf3/12 dcKOs persists at later postnatal stages. At P21, reduction of Plp1 expression in dcKOs compared to controls is still observed (A-B,O). At this stage, protein expression for myelin markers, CNPase and MBP are also reduced in the cortex of dcKOs compared to controls (C-F,O). Note that dcKOs still show expression of neurofilament (NFM) despite the reduction of MBP expression (E-F). As observed in P7, Olig2 is reduced in dcKOs at P21 compared to controls (G-H,O) and Pdgfrα+ cell numbers remain similar between the different genotypes (I-J,O). At P21, Nkx2.2+ cells are similar in controls and dcKOs (K-L,O). To determine if mOL populations continue to improve with age, Plp1 was examined at P60 and remains reduced in dcKOs compared to controls (M-O). Quantification for respective stains counted in the corpus callosum (O). Scale bar in B and N = 1 mm (for A-B, M-N, respectively), D = 200 μm (C-F) and L = 100 μm (for G-L). CC= Corpus Callosum
Embryonic Tcf3/12 dcKOs show ventral progenitor abnormalities
Upon analyzing Olig2 expression and the OPC phenotype in dcKOs at E15.5, it became apparent that dcKOs also exhibit a ventral morphological phenotype observed by Olig2 expression in MGE progenitors (see asterisk Fig. 3E-F and higher magnification images in Fig. 6E-F). The area of the MGE is dramatically increased in the dcKOs compared to controls (120.2% increase in area of Nkx2.1 expression, p < 0.0001, Fig. 6A-K). To better understand this phenotype in the ventral progenitors in Tcf3/12 dcKOs and characterize the increased MGE region, we analyzed the E15.5 stage for patterning and proliferation markers since dcKO MGE phenotype largely resolves by E18.5 (Fig. 3) and that OPCs are actively generated from the MGE and LGE around the E15.5 stage [66]. Therefore, we could get an assessment of general patterning and progenitors in the dcKO. To accomplish this, we analyzed Gsx1, Olig2, and Ascl1 progenitor genes that are expressed in VZ/SVZ cells of the MGE and LGE. In addition, we examined Gsx2, which shows high expression in the LGE and low expression in the MGE and Nkx2.1, which shows specific expression in the entire MGE to address regional patterning in the dcKOs. The expression of these developmental markers reveals that the MGE is expanded in the dcKOs but the overall patterning in the ventral telencephalon is similar to controls suggesting no obvious patterning defect despite the enlarged MGE (Fig. 6A-J). We next examined cell proliferation in MGE progenitor cells at E15.5 to address a stage where the Tcf3/12 dcKO embryos have severely reduced OPC markers and expanded MGE region. Pregnant mice were injected with BrdU 30 min before E15.5 embryonic dissection to identify cells in S-phase. BrdU expression in the VZ of the MGE show similar numbers of proliferating cells per area in controls and dcKOs, with the area of the VZ being larger in the dcKOs (Fig. 6L-Q). This suggests that while dcKOs have more BrdU + cells in the VZ due to the enlarged MGE, the similar numbers of BrdU + cells per area (controlling for size) may indicate the VZ cells are not proliferating at a greater rate at the E15.5 stage. When analyzing the SVZ directly below the VZ at this stage, however, the number of proliferating cells/mm2 is significantly decreased with the dcKOs showing a 42.0% decrease compared to controls (Fig. 6L-Q, p < 0.0001). Therefore, even though the MGE region is larger at E15.5 in dcKOs, defects exist in the number of progenitors proliferating in the secondary progenitor area (SVZ). In addition, this data shows the enlarged MGE SVZ/Mantle region in dcKOs is not occupied by an increase in proliferating cells.

Loss of Tcf3/12 in the ventral telencephalon results in an increased MGE progenitor region and does not impact regional patterning markers. Multiple markers were used to investigate the different progenitor populations within the MGE: Gsx1 is expressed in progenitors in the VZ in the MGE and LGE (A-B), Gsx2 in a high dorsal to low ventral gradient in LGE/MGE progenitors (C-D), Olig2 in LGE/MGE progenitors (E-F), Ascl1 in LGE/MGE progenitors in the VZ and SVZ (G-H), and Nkx2.1 is expressed in progenitors defining MGE boundaries (I-J). Expression of all MGE progenitor populations examined show expression spanning the larger MGE in the dcKOs, while general patterning is maintained. MGE area quantified by area of Nkx2.1 expression (K). BrdU pulses were performed 30 minutes before E15.5 dissection (L-M). Boxes in L and M refer to approximate areas for high magnification shown in N and O. There was no difference in the number of BrdU+ cells in the VZ when controlling for MGE size, but a decrease in BrdU+ cells in the SVZ of the MGE (N-O, quantification P,Q). Scale bars M = 200 μm (for A-M), O= 100 μm (for N-O). MGE = medial ganglionic eminence, LGE = lateral ganglionic eminence, STM = Striatum
To determine if the expanded Nkx2.1 MGE domain in Tcf3/12 dcKOs results in neuronal defects, we analyzed embryonic markers of cortical and striatal interneurons from the MGE. Lhx6 is a downstream target of Nkx2.1 in the MGE and is expressed in developing interneurons [28, 67,68,69,70]. Lhx6 is expressed in the enlarged MGE region of the dcKOs at E15.5 (Fig. 7A, B). Interestingly, dcKOs also show increased Lhx6 staining in the SVZ of the LGE and cortex at E18.5 (Fig. 7C, D), along the path the interneurons use to migrate to the cortex [69]. Moreover, E18.5 dcKOs also showed expanded ventral progenitor domain of Nkx2.1 (compare arrows in Fig. 7E, F) and a 19.9% increase of Nkx2.1 + striatal interneurons compared to controls (p = 0.013, Fig. 7E’,F’,G). Interestingly, the expanded staining for Lhx6 and Nkx2.1 along the migratory path in the LGE region at E18.5 was not apparent earlier at E15.5 (Figs. 6 and 7). Our data show that loss of Tcf3/12 increases the area of the MGE progenitor domain which subsequently leads to increased Lhx6 and Nkx2.1 + cells near the SVZ of the LGE and also increased Nkx2.1 + striatal interneurons. Combined our data suggests that Tcf3 and Tcf12 play a key role in regulating the interneuron oligodendrocyte fate choice in the embryonic ventral telencephalon.

Loss of Tcf3/12 in the ventral telencephalon results in increased neuronal markers. At E15.5 and E18.5, Lhx6 expression was examined in the ventral telencephalon to study cells committed to the interneuron fates (A-D). The increased MGE in the dcKOs at E15.5 is occupied with Lhx6 expression (B). At E18.5, the dcKOs continue to show increased Lhx6 expression in the MGE remnant and along path that the interneurons use to migrate to the cortex in the SVZ of the LGE (C-D). Striatal interneurons, determined by Nkx2.1 expression, shows increased numbers in the dcKOs compared to controls at E18.5 (E-F', Quantification G). Boxes in E and F represent approximate high magnification areas in E’ and F’. Scale bars B = 500 μm (for A-B), D = 500 μm (for C-D), F = 500 μm (for E-F), F’ = 200 μm (for E’-F’). MGE = medial ganglionic eminence, LGE = lateral ganglionic eminence, CTX = Cortex, STM = Striatum
Tcf4 expression is preserved in the dcKOs
Our conditional knockout approach deletes Tcf3 and Tcf12 in both VZ progenitors in the ventral telencephalon and oligodendrocyte lineage. However, the remaining E-protein gene, Tcf4, could compensate for the loss of Tcf3/12. In fact, a recent study has revealed that Tcf4 is expressed in maturing OLs and plays a key role for the appropriate timing of OL differentiation [38]. Therefore, we analyzed Tcf4 protein expression in embryonic and postnatal dcKOs. Tcf4 is enriched in the MGE and ventral telencephalon at E15.5 (Figs. 1E and 8A). In line with the expanded MGE region, Tcf3/12 dcKOs show an 84.2% increase of Tcf4 expression in the MGE (p = 0.0006, Fig. 8A-C). However, there is no difference in Tcf4 expression in the controls and dcKOs when controlling for the increased MGE size in dcKOs, (p = 0.11, Fig. 8A-C). Therefore, the pattern of Tcf4 expression is similar in controls and dcKOs. We next analyzed the oligodendrocyte-rich corpus callosum region at postnatal stages. There is a 29.9% decrease of Tcf4 + cells in the corpus callosum at P21 in the dcKOs (p = 0.0365, Fig. 8D-F). However, there is a decrease of OLs in the corpus callosum (Fig. 5) and therefore less total OLs cells in the area. Thus, the percentage of Olig2 + cells in the corpus callosum in the dcKOs that are positive for Tcf4 is the same between controls and dcKOs (p = 0.69, Fig. 8D-F). There was no difference in Tcf4 expression detected between controls and dcKOs in the postnatal cortex (p = 0.74, Fig. 8G-I) either. Our data indicates that Tcf4 remains expressed in the MGE and oligodendrocyte lineage of Tcf3/12 dcKO and is unable to fully compensate for the loss of Tcf3 and Tcf12 even if it has any redundant role.

Tcf4 is expressed in Tcf3/12 dcKOs. At E15.5, Tcf4 is expressed within the MGE in controls (A). This expression is also seen in the dcKOs, although more Tcf4 expression is seen due to increased MGE size (B). However, when controlling for size of the MGE, there is no difference in Tcf4 expression (Quantification for total intensity and intensity normalized to area - C). At P21, less Tcf4+ cells are detected in the corpus callosum in dcKOs compared to controls (D-E). Since there are overall fewer OLs in the corpus callosum, Tcf4 expression was normalized to percentage of Olig2+ cells co-expressing Tcf4. Once normalized, Tcf4 expression had no significant difference between controls and dcKOs (Quantification for average cell numbers and normalized percentage – F). Tcf4 is highly expressed in the cortex under normal conditions, and no difference is observed between controls and dcKOs (G-H, Quantification – I). Scale bars B = 200 μm (for A-B), E = 100 μm (for D-E), and H= 100 μm (for G-H)
Conditional deletion of Tcf3/12 using Olig1Cre/+ leads to OL lineage defects
We are unable to distinguish if the embryonic OPC or postnatal OL phenotypes in the Tcf3/12 dcKOs is related to the ventral progenitor or interneuron phenotype observed in the dcKOs or if Tcf3 and Tcf12 have a specific role within the OL lineage. To address this, we generated dcKOs with Olig1Cre/+, which recombines later then to Olig2Cre/+ (Fig. 9A-B) in the ventral telencephalon. Unlike Olig2Cre/+ that drives robust recombination in nearly all MGE progenitors and some LGE progenitors [48], Olig1Cre/+ does not robustly target these cells and is highly enriched in OPCs and the subsequent OL lineage cells [17]. Tcf3/12 dcKOs generated with Olig1Cre/+ do not exhibit the abnormal ventral MGE phenotype as Nkx2.1 staining is similar in controls and dcKOs at E15.5 (p = 0.9557, Fig. 9C-E). Nevertheless, there remains a decrease in the OPC markers Olig2 (28.6%, p = 0.0217), Pdgfrα (75.7%, p < 0.0001), and Sox10 (65.0%, p < 0.0001) in the Olig1Cre/+ driven dcKOs (Fig. 9F-N). In addition, Tcf3/12 dcKOs generated with Olig1Cre/+ display reductions in mature OLs as labeled by Plp1 expression similar to the dcKOs generated with Olig2Cre/+. The Olig1Cre/+ dcKOs have 64.0% less Plp1 + cells compared to controls at P21 (p < 0.0001, Fig. 9O-Q). This indicates that Tcf3 and Tcf12 play a role in embryonic OPC generation and postnatal OL differentiation that is independent of the MGE phenotypes observed in Tcf3/12 dcKOs generated with Olig2Cre/+. Collectively, our data suggests that these two E-protein genes play multiple roles during development in the generation of appropriate numbers of OPCs and cortical interneurons in the embryonic telencephalon and the differentiation of OLs after birth.

Conditional knock out of Tcf3/12 using Olig1cre/+results in similar oligodendrocyte defects, without the MGE phenotype observed in Tcf3/12 dcKOs generated with Olig2Cre/+. Fate map of Olig2Cre/+using Rosatom/+as a reporter of Cre activation shows recombination throughout the MGE and LGE and cells migrating to the cortex (A). Fate map of Olig1Cre/+using Rosatom/+ shows later recombination in the ventral progenitors and is enriched in OPCs (B). Olig1Cre/+ driven conditional knockout Tcf3/12 preserves MGE size in the dcKOs, as marked by Nkx2.1 expression (C-E). Similar to Tcf3/12 dcKOs generated with Olig2Cre/+, Tcf3/12 dcKOs generated with Olig1Cre/+have a reduction of Olig2 expression in the parenchyma compared to controls at E15.5 (F-H). Tcf3/12 dcKOs generated with Olig1Cre/+also have a dramatic reduction in Pdgfrα+ and Sox10+ OPCs compared to controls at E15.5 (I-K and L-N, respectively) and reduced numbers of Plp+ cells compared to controls at P21, (O-Q).Scale bars M= 500 μm (for A-M) and P= 1 mm (for O-P)
Discussion
This study examines the role of Tcf3 and Tcf12 during OL development. Our conditional loss-of-function studies reveals that Tcf3 and Tcf12 play roles in the development of ventral progenitors in the MGE by regulating the interneuron and OPC fate choice, and also in the OL lineage for the generation of embryonic OPCs and the appropriate numbers of maturing postnatal OLs. These phenotypes are observed even though the remaining E-protein, Tcf4, continues to be expressed in the embryonic (MGE) and postnatal (OLs) populations effected by Tcf3/12 loss. Moreover, we found the reduction of embryonic OPCs and postnatal OLs in dcKOs occurs in the absence of the MGE phenotype by using a conditional knockout approach to enrich for deletion in the OPC/OL lineage using Olig1cre/+. Overall, data from our study uncover the importance for Tcf3 and Tcf12 in progenitors of the ventral telencephalon and during oligodendrogenesis.
The Tcf3/12 dcKOs show a dramatic phenotype in the loss of OPCs labeled by Sox10 and Pdgfrα in the telencephalon during embryonic development. In line with this, loss of class II bHLH TFs Ascl1 and Olig2, potential binding partners for E-protein, results in severe reduction of embryonic OPCs [11, 12, 58, 71]. One possible explanation for the loss of OPCs in the dcKOs could be a result of disrupting the class II function in OPC production by removal of the class I bHLH binding partners (Tcf3 and Tcf12). Recent biochemical studies revealed that Tcf4 has interaction with Olig1, Olig2, and Ascl1, Tcf12 interacted with Olig1 and Ascl1, and Tcf3 interacted with Olig2 and Ascl1 [38]. It is striking that the OPC phenotype in our model is unique from studies of Tcf4 KOs that show no perturbations of OPCs in the spinal cord or the ex vivo telencephalon cultures but robust defects in OL differentiation [38]. Therefore, it seems possible that combined Tcf3 and Tcf12 function in OPC generation during early stages where as Tcf4 functions to regulate the timing of OL differentiation at later stages. Another recent study showed enhanced survival of OPCs that migrate to the olfactory bulb in Tcf4 conditional mutants generated with Nkx2.1-cre suggesting there also may be region specific effects in the CNS for E-protein loss in the CNS [72]. However, our study revealed that Tcf4 remains expressed in the ventral regions of Tcf3/12 dcKOs suggesting its inability to fully compensate for the loss of Tcf3 and Tcf12 during OPC generation even if Tcf4 is involved in OPC survival.
Tcf3−/− and Tcf12−/− mice showed similar lethal phenotypes to Tcf3/12+/− mice, suggesting E-protein dosage is more important than E-protein identity [43]. Importance of bHLH gene dosage has also been demonstrated in OPCs, as Ascl1/Olig2+/− show more severe phenotypes than single Ascl1+/− and Olig2+/− mice [12]. Also, Tcf4/Olig2+/− showed similar OL differentiation phenotypes to Tcf4−/− which suggests a key interaction of Tcf4 and Olig2 in OLs [38]. In our experimental approach we conditionally deleted all four alleles of Tcf3/12. In addition, both Olig2Cre/+ and Olig1 Cre/+ are knock-in knock-out Cre driver mice, so only one functional allele for the gene is present, further reducing the bHLH TF dosage in our Tcf3/12 dcKOs. Given the recent finding that Tcf12-Olig1 and Tcf3-Olig2 interaction partners exist [38], future studies are required to understand whether the phenotypes in the Tcf3/12 dcKOs are due to compound dosage effects specifically with Olig1 or Olig2 or if Tcf3/12 are distinctly required in embryonic production of OPCs.
The enlarged MGE in the Tcf3/12 dcKO is a phenotype that has not been reported in any other bHLH knockout model. In fact, loss of Ascl1 results in the opposite phenotype with reduced MGE progenitors and a near complete absence of a pronounced ventral most eminence region [73]. This suggests that the MGE phenotype in Tcf3/12 dcKO is not mediated by reduced Ascl1 function. Interestingly, even with this enlarged MGE region, the general patterning of the ventral telencephalon in Tcf3/12 dcKOs appears to be preserved through normal regional expression of different progenitor markers. However, the generation of appropriate glia and neurons from MGE was severely disrupted in dcKOs with nearly a complete absence of OPCs and an expansion of Nkx2.1 and Lhx6 expression into the LGE, which is likely the result of the increased number of MGE cells migrating in a tangential manner into the LGE. While the increased MGE phenotype is unique to our Tcf3/12 dcKOs, other bHLH knockout models have cortical interneuron phenotypes. Ascl1 is required for cortical interneuron production and overexpression promotes cortical interneuron migration [73, 74]. It is possible that increased total Ascl1 + cells in the enlarged MGE of dcKOs produces a gain-of-function effect of Ascl1, leading to increase migration of the expanded MGE cells. Alternatively, Olig1−/− mice have been shown to overproduce interneurons at the expense of oligodendrocytes from the medial septal progenitor regions and ventral most MGE [62]. The role of Olig1 in regulating cell fate is consistent with the observations in Tcf3/12 dcKOs, suggesting that loss of Tcf3/12 could produce a loss-of-function for Olig1. In fact, Tcf12 was previously identified to be Olig1’s preferred binding partner, while Tcf3 also had some ability to heterodimerize with Olig1 [38]. However, it likely Olig1 is not alone in mediating all the Tcf3/12 dcKO phenotypes, since the Olig1−/− mice do not show the enlarged MGE phenotype or the severe embryonic OPC phenotype in the ventral telencephalon observed in the Tcf3/12 dcKOs [58, 62]. Olig2−/− embryos have a very severe OPC generation phenotype in the ventral telencephalon similar to the Tcf3/12 dcKOs [58, 60, 71]. However, key phenotypic differences remain including that Olig2−/− embryos do not display the enlarged MGE and show minimal defects in cortical interneuron development from the MGE with only a specific reduction in ChAT + cells in the ventral regions [75,76,77]. Also, the presence of Olig2 + cells in dcKOs although reduced and the severe reduction of Pdgfrα and Sox10 cells suggests that there may be a defect in OPC expansion or maturation. The similarities between Olig1 KO, Olig2 KO, and Tcf3/12 dcKO defects in oligodendrogenesis, suggest that Tcf3, Tcf12 and Olig1/2 share some common roles during development. Future experiments are required to understand whether there is an in vivo interaction between Tcf3/12 and Olig1/2 or whether these bHLH TFs converge on the same developmental event.
It is not uncommon to observe recovery of OL populations in genetic models that initially perturb these populations [12, 13, 21, 58]. The timing of recovery provides some insight into the Tcf3/12 dcKO phenotype. OPC production in the telencephalon has distinct spatiotemporal components. OPCs initially are produced by the MGE around E12.5, then are produced by both the MGE and LGE around E15.5, and OPCs generated after E17.5 are produced from the dorsal telencephalon [66, 78]. It is generally accepted that the cortically derived OPCs will replace the majority of MGE derived OPCs to populate the telencephalon [66]. The timing of the OPC recovery observed in this study would be consistent with the production of cortical/dorsal OPCs, indicating Tcf3 and Tcf12 may not play an important role in cortical OPC generation unlike ventral (LGE/MGE) OPC generation. However, even after OPC recovery at P7, we observed sustained reductions in the mature OL populations of Tcf3/12 dcKOs at P7, P21, and P60. Therefore, the recovered OPCs as early as P7 in the dcKOs, appear to be incapable of properly differentiating as late as P60, suggesting that Tcf3 and Tcf12 may also be involved in the differentiation of OLs at postnatal stages. In support of this idea, our later recombination studies of Tcf3/12 using Olig1Cre/+ did not result in an expanded MGE region but still caused a reduction in embryonic OPCs and also mOLs at postnatal stages. This suggests that that Tcf3 and Tcf12 also play distinct roles in early embryonic OPC generation and later OL differentiation at postnatal stages.
Our results reveal that Tcf3 and Tcf12 play a role in regulating cell fate decisions from the MGE, generating embryonic OPCs, and generating appropriate numbers of OLs at postnatal stages. There remains much that is not understood about class I bHLH TFs and their roles or interactions with class II bHLH TFs. However, this study and the recent work on Tcf4 [38, 72]. have implicated all 3 E-proteins as major regulators during distinct stages of developmental oligodendrogenesis.
Availability of data and materials
Original data for this study are included in the article. Inquires for data can be directed to the corresponding author.
References
Bradl M, Lassmann H. Oligodendrocytes: biology and pathology. Acta Neuropathol. 2010;119:37–53.
Kuhn S, Gritti L, Crooks D, Dombrowski Y. Oligodendrocytes in development, myelin Generation and Beyond. Cells. 2019;8: 1424.
Emery B, Lu QR. Transcriptional and epigenetic regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb Perspect Biol. 2015;7: a020461.
Elbaz B, Popko B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019;42:263–77.
Marques S, Van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Väremo L, Giacomello S, Falcão AM, Meijer M, Björklund ÃK, Hjerling-Leffler J, Taft RJ, Castelo-Branco G. Transcriptional convergence of Oligodendrocyte lineage progenitors during development. Dev Cell. 2018;46:504-517e507.
Weng Q, et al. Single-cell Transcriptomics uncovers glial progenitor diversity and cell fate determinants during development and gliomagenesis. Cell Stem Cell. 2019;24:707-723e708.
Li X, Liu G, Yang L, Li Z, Zhang Z, Xu Z, Cai Y, Du H, Su Z, Wang Z, Duan Y, Chen H, Shang Z, You Y, Zhang Q, He M, Chen B, Yang Z. Decoding cortical glial cell development. Neurosci Bull. 2021;37:440–60.
Zhou Q, Wang S, Anderson DJ. Identification of a Novel Family of Oligodendrocyte Lineage-Specific Basic Helix–Loop–Helix transcription factors. Neuron. 2000;25:331–43.
Lu QR, Yuk D-I, Alberta JA, Zhu Z, Pawlitzky I, Chan J, Mcmahon AP, Stiles CD, Rowitch DH. Sonic hedgehog–regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian Central Nervous System. Neuron. 2000;25:317–29.
Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73.
Yue T, Xian K, Hurlock E, Xin M, Kernie SG, Parada LF, Lu QR. A critical role for dorsal progenitors in cortical myelination. J Neurosci. 2006;26:1275–80.
Parras CM, Hunt C, Sugimori M, Nakafuku M, Rowitch D, Guillemot F. The Proneural Gene Mash1 specifies an early Population of Telencephalic Oligodendrocytes. J Neurosci. 2007;27:4233–42.
Sugimori M, Nagao M, Bertrand N, Parras CM, Guillemot FO, Nakafuku M. Combinatorial actions of patterning and HLH transcription factors in the spatiotemporal control of neurogenesis and gliogenesis in the developing spinal cord. Development. 2007;134:1617–29.
Zhu X, Zuo H, Maher BJ, Serwanski DR, Loturco JJ, Lu QR, Nishiyama A. Olig2-dependent developmental fate switch of NG2 cells. Development. 2012;139:2299–307.
Mei F, Wang H, Liu S, Niu J, Wang L, He Y, Etxeberria A, Chan JR, Xiao L. Stage-specific deletion of Olig2 conveys opposing functions on differentiation and maturation of oligodendrocytes. J Neurosci. 2013;33:8454–62.
Parras CM, Galli R, Britz O, Soares S, Galichet C, Battiste J, Johnson JE, Nakafuku M, Vescovi A, Guillemot F. Mash1 specifies neurons and oligodendrocytes in the postnatal brain. EMBO J. 2004;23:4495–505.
Xin M, Yue T, Ma Z, Wu FF, Gow A, Lu QR. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci. 2005;25:1354–65.
Sugimori M, Nagao M, Parras CM, Nakatani H, Lebel ML, Guillemot FO, Nakafuku M. Ascl1 is required for oligodendrocyte development in the spinal cord. Development. 2008;135:1271–81.
Nakatani H, Martin E, Hassani H, Clavairoly A, Maire CL, Viadieu A, Kerninon C, Delmasure A, Frah M, Weber M, Nakafuku M, Zalc B, Thomas J-L, Guillemot F, Nait-Oumesmar B, Parras C. Ascl1/Mash1 promotes brain oligodendrogenesis during myelination and remyelination. J Neurosci. 2013;33:9752–68.
Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LM, Mao M, Chan JR, Wu J, Lu QR. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–61.
Dai J, Bercury KK, Ahrendsen JT, Macklin WB. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J Neurosci. 2015;35:4386–402.
Murre C, Mccaw PS, Vaessin H, Caudy M, Jan LY, Jan YN, Cabrera CV, Buskin JN, Hauschka SD, Lassar AB, Weintraub H, Baltimore D. Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell. 1989;58:537–44.
Murre C, Bain G, Van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, Quong MW, Rivera RR, Stuiver MH. Structure and function of helix-loop-helix proteins. Biochim et Biophys Acta (BBA) - Gene Struct Expression. 1994;1218:129–35.
Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–40.
Chen T, Wu Q, Zhang Y, Lu T, Yue W, Zhang D. Tcf4 controls neuronal Migration of the cerebral cortex through regulation of Bmp7. Front Mol Neurosci. 2016;9:94.
Jung M, Häberle BM, Tschaikowsky T, Wittmann MT, Balta EA, Stadler VC, Zweier C, Dörfler A, Gloeckner CJ, Lie DC. Analysis of the expression pattern of the schizophrenia-risk and intellectual disability gene TCF4 in the developing and adult brain suggests a role in development and plasticity of cortical and hippocampal neurons. Mol Autism. 2018;9:20.
Long JE, Cobos I, Potter GB, Rubenstein JL. Dlx1&2 and Mash1 transcription factors control MGE and CGE patterning and differentiation through parallel and overlapping pathways. Cereb Cortex. 2009;19(Suppl 1):i96-106.
Sandberg M, Taher L, Hu J, Black BL, Nord AS, Rubenstein JLR. Genomic analysis of transcriptional networks directing progression of cell states during MGE development. Neural Dev. 2018;13:21.
Sussman CR, Davies JE, Miller RH. Extracellular and intracellular regulation of oligodendrocyte development: roles of sonic hedgehog and expression of E proteins. Glia. 2002;40:55–64.
Gokhan S, Marin-Husstege M, Yung SY, Fontanez D, Casaccia-Bonnefil P, Mehler MF. Combinatorial profiles of oligodendrocyte-selective classes of transcriptional regulators differentially modulate myelin basic protein gene expression. J Neurosci. 2005;25:8311–21.
Wang J, Pol SU, Haberman AK, Wang C, O’Bara MA, Sim FJ. Transcription factor induction of human oligodendrocyte progenitor fate and differentiation. Proceedings of the National Academy of Sciences. 2014;111:E2885–94.
Chen H-Y, Bohlen JF, Maher BJ. Molecular and Cellular function of transcription factor 4 in Pitt-Hopkins Syndrome. Dev Neurosci. 2021;43:159–67.
Brockschmidt A, Todt U, Ryu S, Hoischen A, Landwehr C, Birnbaum S, Frenck W, Radlwimmer B, Lichter P, Engels H, Driever W, Kubisch C, Weber RG. Severe mental retardation with breathing abnormalities (Pitt–Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum Mol Genet. 2007;16:1488–94.
Li H, Zhu Y, Morozov YM, Chen X, Page SC, Rannals MD, Maher BJ, Rakic P. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol Psychiatry. 2019;24:1235–46.
Mesman S, Bakker R, Smidt MP. Tcf4 is required for correct brain development during embryogenesis. Mol Cell Neurosci. 2020;106: 103502.
Wang Y, Lu Z, Zhang Y, Cai Y, Yun D, Tang T, Cai Z, Wang C, Fang F, Yang Z, Behnisch T, Xie Y. Transcription factor 4 safeguards hippocampal dentate Gyrus Development by regulating neural progenitor Migration. Cereb Cortex. 2020;30:3102–15.
Phan BN, et al. A myelin-related transcriptomic profile is shared by Pitt–Hopkins syndrome models and human autism spectrum disorder. Nat Neurosci. 2020;23:375–85.
Wedel M, Fröb F, Elsesser O, Wittmann M-T, Lie C, Reis D, Wegner A. Transcription factor Tcf4 is the preferred heterodimerization partner for Olig2 in oligodendrocytes and required for differentiation. Nucleic Acids Res. 2020;48:4839–57.
Zhuang Y, Barndt RJ, Pan L, Kelley R, Dai M. Functional replacement of the mouse E2A gene with a human HEB cDNA. Mol Cell Biol. 1998;18:3340–9.
Barndt R, Dai MF, Zhuang Y. A novel role for HEB downstream or parallel to the pre-TCR signaling pathway during alpha beta thymopoiesis. J Immunol. 1999;163:3331–43.
Barndt RJ, Dai M, Zhuang Y. Functions of E2A-HEB heterodimers in T-Cell development revealed by a Dominant negative mutation of HEB. Mol Cell Biol. 2000;20:6677–85.
Wojciechowski J, Lai A, Kondo M, Zhuang Y. E2A and HEB are required to block thymocyte proliferation prior to pre-TCR expression. J Immunol. 2007;178:5717–26.
Ravanpay AC, Olson JM. E protein dosage influences brain development more than family member identity. J Neurosci Res. 2008;86:1472–81.
Dessaud E, Yang LL, Hill K, Cox B, Ulloa F, Ribeiro A, Mynett A, Novitch BG, Briscoe J. Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature. 2007;450:717–20.
Waclaw RR, Allen ZJ, Bell SM, Erdélyi F, Szabó G, Potter SS, Campbell K. The zinc finger transcription factor Sp8 regulates the generation and diversity of olfactory bulb interneurons. Neuron. 2006;49:503–16.
Waclaw RR, Ehrman LA, Pierani A, Campbell K. Developmental Origin of the neuronal subtypes that comprise the Amygdalar Fear Circuit in the mouse. J Neurosci. 2010;30:6944–53.
Ehrman LA, Nardini D, Ehrman S, Rizvi TA, Gulick J, Krenz M, Dasgupta B, Robbins J, Ratner N, Nakafuku M, Waclaw RR. The protein tyrosine phosphatase Shp2 is required for the generation of oligodendrocyte progenitor cells and myelination in the mouse Telencephalon. J Neurosci. 2014;34:3767–78.
Qin S, Ware SM, Waclaw RR, Campbell K. Septal contributions to olfactory bulb interneuron diversity in the embryonic mouse telencephalon: role of the homeobox gene Gsx2. Neural Dev. 2017;12(1):13.
Toresson H, Potter SS, Campbell K. Genetic control of dorsal-ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development. 2000;127:4361–71.
Kohli V, Nardini D, Ehrman LA, Waclaw RR. Characterization ofGlcci1expression in a subpopulation of lateral ganglionic eminence progenitors in the mouse telencephalon. Dev Dyn. 2018;247:222–8.
Sandberg M, Flandin P, Silberberg S, Su-Feher L, Price JD, Hu JS, Kim C, Visel A, Nord AS, Rubenstein JLR. Transcriptional networks controlled by NKX2-1 in the development of Forebrain GABAergic neurons. Neuron. 2016;91:1260–75.
Roberts VJ, Steenbergen R, Murre C. Localization of E2A mRNA expression in developing and adult rat tissues. Proc Natl Acad Sci U S A. 1993;90:7583–7.
Pan L, Hanrahan J, Li J, Hale LP, Zhuang Y. An analysis of T cell intrinsic roles of E2A by conditional gene disruption in the Thymus. J Immunol. 2002;168:3923–32.
Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol. 1996;16:2898–905.
Pringle NP, Richardson WD. A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development. 1993;117:525–33.
Takebayashi H, Yoshida S, Sugimori M, Kosako H, Kominami R, Nakafuku M, Nabeshima Y. Dynamic expression of basic helix-loop-helix olig family members: implication of Olig2 in neuron and oligodendrocyte differentiation and identification of a new member, Olig3. Mech Dev. 2000;99:143–8.
Tekki-Kessaris N, Woodruff R, Hall AC, Gaffield W, Kimura S, Stiles CD, Rowitch DH, Richardson WD. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development. 2001;128:2545–54.
Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for olig function indicates a motor Neuron/Oligodendrocyte connection. Cell. 2002;109:75–86.
Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–70.
Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y-I. The Basic Helix-Loop-Helix factor Olig2 is essential for the development of Motoneuron and Oligodendrocyte Lineages. Curr Biol. 2002;12:1157–63.
Küspert M, Hammer A, Bösl MR, Wegner M. Olig2 regulates Sox10 expression in oligodendrocyte precursors through an evolutionary conserved distal enhancer. Nucleic Acids Res. 2011;39:1280–93.
Silbereis C, John, Nobuta H, Tsai H-H, Heine M, Vivi, Mckinsey L, Gabriel, Meijer H, Dimphna HA, Mackenzie, Petryniak A, Magda, Potter B, Gregory AA, John, Baraban C, Scott SD, Charles, Rubenstein LR, John, Rowitch H. Olig1 function is required to repress Dlx1/2 and Interneuron production in mammalian brain. Neuron. 2014;81:574–87.
Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–22.
Cai J, Zhu Q, Zheng K, Li H, Qi Y, Cao Q, Qiu M. Co-localization of Nkx6.2 and Nkx2.2 homeodomain proteins in differentiated myelinating oligodendrocytes. Glia. 2010;58:458–68.
Cai J, Chen Y, Cai WH, Hurlock EC, Wu H, Kernie SG, Parada LF, Lu QR. A crucial role for Olig2 in white matter astrocyte development. Development. 2007;134:1887–99.
Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–9.
Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–8.
Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–70.
Marín O, Anderson SA, Rubenstein JLR. Origin and molecular specification of Striatal Interneurons. J Neurosci. 2000;20:6063–76.
Vogt D, Hunt RF, Mandal S, Sandberg M, Silberberg SN, Nagasawa T, Yang Z, Baraban SC, Rubenstein JL. Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron. 2014;82:350–64.
Chapman H, Riesenberg A, Ehrman LA, Kohli V, Nardini D, Nakafuku M, Campbell K, Waclaw RR. Gsx transcription factors control neuronal versus glial specification in ventricular zone progenitors of the mouse lateral ganglionic eminence. Dev Biol. 2018;442:115–26.
Zhang Y, Cai Y, Wang Y, Deng X, Zhao Y, Zhang Y, Xie Y. Survival control of oligodendrocyte progenitor cells requires the transcription factor 4 during olfactory bulb development. Cell Death Dis. 2021;12(1):91.
Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–34.
Liu Y-H, Tsai J-W, Chen J-L, Yang W-S, Chang P-C, Cheng P-L, Turner DL, Yanagawa Y, Wang T-W, Yu J-Y. Ascl1 promotes tangential migration and confines migratory routes by induction of Ephb2 in the telencephalon. Sci Rep. 2017;7: 42895.
Furusho M, Ono K, Takebayashi H, Masahira N, Kagawa T, Ikeda K, Ikenaka K. Involvement of the Olig2 transcription factor in cholinergic neuron development of the basal forebrain. Dev Biol. 2006;293:348–57.
Petryniak MA, Potter GB, Rowitch DH, Rubenstein JLR. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell Fate Acquisition in the developing forebrain. Neuron. 2007;55:417–33.
Ono K, Takebayashi H, Ikeda K, Furusho M, Nishizawa T, Watanabe K, Ikenaka K. Regional- and temporal-dependent changes in the differentiation of Olig2 progenitors in the forebrain, and the impact on astrocyte development in the dorsal pallium. Dev Biol. 2008;320:456–68.
Winkler CC, Yabut OR, Fregoso SP, Gomez HG, Dwyer BE, Pleasure SJ, Franco SJ. The dorsal Wave of Neocortical Oligodendrogenesis begins embryonically and requires multiple sources of Sonic hedgehog. J Neurosci. 2018;38:5237–50.
Acknowledgements
We would like to thank Drs. Kenneth Campbell and Masato Nakafuku (CCHMC) for critical reading or comments on data analysis for this manuscript.
Funding
This work was supported by NIH grant NS088529 (RW).
Author information
Authors and Affiliations
Contributions
Author Contributions Mary Jo Talley performed experiments, data analysis, figure generation, first draft of the manuscript, and edited the manuscript. Diana Nardini provided technical assistance with animal husbandry, histology, and editing the manuscript. Lisa Ehrman provided technical assistance with experimental design, aided in interpretation of the data, and edited the manuscript. Richard Lu provided key research tools for the study, aided in interpretation of the data, and edited the manuscript. Ronald Waclaw conceived of the project, designed and supervised the experiments, and contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Statement in methods section: Protocols for animal experiments using mice were approved by the Institutional Animal Care and Use Committee at the Cincinnati Children’s Hospital Medical Center and carried out in accordance with National Institutes of Health guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Talley, M.J., Nardini, D., Ehrman, L.A. et al. Distinct requirements for Tcf3 and Tcf12 during oligodendrocyte development in the mouse telencephalon. Neural Dev 18, 5 (2023). https://doi.org/10.1186/s13064-023-00173-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13064-023-00173-z