
Abstract
Background
Wolf-Hirschhorn syndrome (WHS) is a well-defined disorder, whose core phenotype encompasses growth restriction, facial gestalt, intellectual disability and seizures. Nevertheless, great phenotypic variability exists due to the variable extent of the responsible 4p deletion. In addition, exome sequencing analyses, recently identified two genes, namely NSD2 and NELFA, whose loss-of-function variants contribute to a clinical spectrum consistent with atypical or partial WHS.
The observation of patients exhibiting clinical features resembling WHS, with only mild developmental delay and without the typical dysmorphic features, carrying microdeletions sparing NSD2, has lead to the hypothesis that NSD2 is responsible for the intellectual disability and the facial gestalt of WHS. While presenting some of the typical findings of WHS (intellectual disability, facial gestalt, microcephaly, growth restriction and congenital heart defects), NSD2-deleted children tend to display a milder spectrum of skeletal abnormalities, usually consisting of clinodactyly, and do not exhibit seizures.
We describe the clinical picture of a child with WHS due to a de novo mutation of NSD2 and discuss the clinical and diagnostic implications.
Case presentation
A 6-year-old boy was evaluated for a history of intrauterine growth restriction, low birth weight, neonatal hypotonia, and psychomotor delay. No episodes of seizure were reported. At physical examination, he displayed marphanoid habitus, muscle hypotrophy and facial dysmorphisms consisting in high frontal hairline, upslanting palpebral fissures and full lips with bifid ugula. Cryptorchidism, shawl scrotum, mild clinodactyly of the right little finger and bilateral syndactyly of the II and III toes with sandal gap were also noted. The radiographic essay demonstrated delayed bone age and echocardiography showed mild mitral prolapse. Whole genome sequencing analysis revealed a heterozygous de novo variant of NSD2 (c.2523delG).
Conclusions
Full WHS phenotype likely arises from the cumulative effect of the combined haploinsufficiency of several causative genes mapping within the 4p16.3 region, as a contiguous genes syndrome, with slightly different phenotypes depending on the specific genes involved in the deletion.
When evaluating children with pictures resembling WHS, in absence of seizures, clinicians should consider this differential diagnosis.
Similar content being viewed by others


Background
Wolf-Hirschhorn syndrome (WHS) is a well-defined disorder due to variable size-deletions of the chromosomal region 4p16.3, characterized by a clinical picture encompassing growth restriction, developmental delay, microcephaly, congenital hypotonia and major malformations, including midline, heart, renal and skeletal defects, along with the typical facial gestalt, consisting of the so called “Greek warrior helmet” appearance (high forehead, continuing to a wide nasal bridge, with short philtrum, high arched eyebrows, hypertelorism, and micrognathia). Seizures occur in nearly all affected patients within the age of 3 years and complicate the management, acting as a significant prognostic factor for the final degree of intellectual disability.
In front of the great phenotypic variability of WHS, depending mostly on the extent of the 4p deletion, the core WHS phenotype is conventionally defined by the association of intellectual disability, growth delay, facial gestalt and seizures [1]. Thus, two minimal critical regions responsible for WHS (WHSCR) have been identified, corresponding to the smallest region, whose haploinsufficiency determines the core phenotype [2,3,4].
More recently, exome sequencing analyses identified two genes within the WHSCR, whose loss-of-function variants contribute to a clinical spectrum consistent with atypical or partial WHS: WHS candidate gene 1 (WHSC1), also known as Nuclear receptor-binding Set Domain-protein 2 (NSD2), contained only partly within the WHSCR [5], and WHS candidate gene 2 (WHSC2), also known as Negative Elongation Factor Complex Member A (NELFA), entirely contained within the WHSCR [6].
We describe a patient with a de novo variant of NSD2 and discuss the clinical implications.
Case presentation
The proband was a 6-year-old boy, born at 34 weeks of gestation by cesarean section from healthy, non-consanguineous parents. Gestation was complicated by intrauterine growth restriction (IUGR) and the baby displayed low birth weight. The neonatal period was characterized by hypotonia, followed by psychomotor delay. No episodes of seizure were reported.
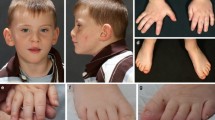
At physical examination, he displayed marphanoid habitus, muscle hypotrophy and facial dysmorphisms consisting in high frontal hairline, upslanting palpebral fissures and full lips with bifid ugula (Fig. 1). Cryptorchidism, shawl scrotum, mild clinodactyly of the right little finger, bilateral syndactyly of the II and III toes with sandal gap and a small café-au-lait spot on dorsum were also noted. The radiographic essay demonstrated delayed bone age and echocardiography showed mild mitral prolapse.

Patient’s facies, characterized by high frontal hairline, upslanting palpebral fissures and full lips
Both single nucleotide polymorphism-arrays and next-generation intellectual disability gene panel proved negative. Whole genome sequencing analysis revealed a heterozygous de novo variant of NSD2 (c.2523delG).
Discussion and conclusions
NSD2 acts as a histone methyltransferase, responsible for the methylation of HEK36, thus explaining the occurrence of developmental delay in carriers of NSD2 variants, in light of the crucial role of histones modification in brain development. Of note, the description of two patients with intact NSD2, exhibiting clinical features resembling WHS but only mild developmental delay [7], has lead to the assumption that the haploinsufficiency of NSD2 is responsible for the developmental delay, typically observed in WHS patients; this hypothesis has been further supported by the documentation of a higher degree of developmental delay in patients with disrupted NSD2, compared with those with the intact gene [8, 9]. Autism spectrum disorder has been reported in eight NSD2-haploinsufficient children [10, 11]. Moreover, deletions of NSD2 are considered responsible for the facial gestalt of WHS, in light of the observation of non-specific findings consistent with WHS (growth and developmental delay) but without the typical dysmorphic features, in several patients with microdeletions sparing NSD2 [12, 13].
Hence, the clinical spectrum of NSD2 deletion encompasses: prenatal and postnatal growth restriction [14], microcephaly, developmental delay [15], congenital heart defects and several phenotypic traits, including hypertelorism, upward-slanting palpebral fissures, prominent nasal bridge, abnormal teething and micrognathia. Cleft palate has been described in fourteen patients [16,17,18,19,20,21]. Compared to WHS patients, NSD2-deleted children tend to display a milder spectrum of skeletal abnormalities, usually consisting of clinodactyly [22]. Table 1 summarizes the previously reported cases of NSD2 haploinsufficiency. Remarkably, seizures are not usually part of the clinical spectrum of NSD2 variants.
LETM1 (Leucine zipper/EF-hand containing transmembrane), involved in calcium signaling and mapping within the WHSCR, had been previously identified as responsible for seizures. However, this assumption has recently been questioned by the observation of the occurrence of seizures in children carrying terminal 4p deletions sparing LETM1, and of the lack of seizure in individuals with interstitial deletions including LETM1, but preserving a relatively large terminal 4p segment [23]: these observations suggest that the haploinsufficiency of LETM1 alone may not be sufficient in causing seizures, which would rather result from the effect of additional candidate genes [24].
Remarkably, the recurrence risk of NSD2 variants is 50% and must be taken into account when counseling families of affected individuals.
In conclusion, full WHS phenotype probably arises from the cumulative effect of the combined haploinsufficiency of several causative genes mapping into the 4p16.3 region, as a contiguous genes syndrome, with slightly different phenotypes depending on the specific genes involved in the deletion [25].
NSD2 haploinsufficiency is responsible of a distinctive entity, with clinical findings falling to some extent within the WHS phenotype, but not sufficient to allow a conclusive diagnosis of WHS.
When evaluating children with pictures resembling WHS, clinicians should bear this condition in mind as a possible differential diagnosis.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- WHS:
-
Wolf-Hirschhorn syndrome
- WHSC1 :
-
Wolf-Hirschhorn syndrome candidate gene 1
- NSD2 :
-
Nuclear receptor-binding Set Domain-protein 2
- WHSC2 :
-
Wolf-Hirschhorn syndrome candidate gene 2
- WHSCR:
-
Wolf-Hirschhorn syndrome Critical Region
- NELFA :
-
Negative Elongation Factor Complex Member A
- LETM1 :
-
Leucine zipper/EF-hand containing transmembrane
References
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, et al. On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literature review. Am J Med Genet C: Semin Med Genet. 2008;148C(4):257–69.
Rauch A, Schellmoser S, Kraus C, Dörr HG, Trautmann U, Altherr MR, et al. First known microdeletion within the Wolf-Hirschhorn syndrome critical region refines genotype-phenotype correlation. Am J Med Genet. 2001;99(4):338–42.
Zollino M, Lecce R, Fischetto R, Murdolo M, Faravelli F, Selicorni A, et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72(3):590–7.
Rodríguez L, Zollino M, Climent S, Mansilla E, López-Grondona F, Martínez-Fernández ML, et al. The new Wolf-Hirschhorn syndrome critical region (WHSCR-2): a description of a second case. Am J Med Genet A. 2005;136(2):175–8.
Derar N, Al-Hassnan ZN, Al-Owain M, Monies D, Abouelhoda M, Meyer BF, et al. De Novo Truncating Variants in WHSC1 Recapitulate the Wolf-Hirschhorn (4p16.3 Microdeletion) Syndrome Phenotype. Genet Med. 2019;21(1):185–8.
Cyr AB, Nimmakayalu M, Longmuir SQ, Patil SR, Keppler-Noreuil K, Shchelochkov OA. A novel 4p16.3 microduplication distal to WHSC1 and WHSC2 characterized by oligonucleotide array with new phenotypic features. Am J Med Genet Part A. 2011;155:2224–8.
South ST, Hannes F, Fisch GS, Vermeesch JR, Zollino M. Pathogenic significance of deletions distal to the currently described Wolf-Hirschhorn syndrome critical regions on 4p16.3. Am J Med Genet Part C. 2008;148C:270–4.
Izumi K, Okuno H, Maeyama K, Sato S, Yamamoto T, Torii C, et al. Interstitial microdeletion of 4p16.3: Contribution of WHSC1 haploinsufficiency to the pathogenesis of developmental delay in Wolf–Hirschhorn syndrome. Am J Med Genet Part A. 2010;152A:1028–32.
Okamoto N, Ohmachi K, Shimada S, Shimojima K, Yamamoto T. 109 kb deletion of chromosome 4p16.3 in a patient with mild phenotype of Wolf-Hirschhorn syndrome. Am J Med Genet A. 2013;161A(6):1465–9.
Barrie ES, Alfaro MP, Pfau RB, Goff MJ, McBride KL, Manickam K, et al. De Novo Loss-Of-Function Variants in NSD2 (WHSC1) Associate With a Subset of Wolf-Hirschhorn Syndrome. Cold Spring Harb Mol Case Stud. 2019;5(4):a004044.
Zanoni P, Steindl K, Sengupta D, Joset P, Bahr A, Sticht H, et al. Loss-of-function and missense variants in NSD2 cause decreased methylation activity and are associated with a distinct developmental phenotype. Genet Med. 2021;23(8):1474–83.
South ST, Bleyl SB, Carey JC. Two unique patients with novel microdeletions in 4p16.3 that exclude the WHS critical regions: Implications for critical region designation. Am J Med Genet Part A. 2007;143A:2137–42.
Engbers H, van der Smagt JJ, van’t Slot R, Vermeesch JR, Hochstenbach R, Poot M. Wolf-Hirschhorn Syndrome Facial Dysmorphic Features in a Patient With a Terminal 4p16.3 Deletion Telomeric to the WHSCR and WHSCR 2 Regions. Eur J Hum Genet. 2009;17(1):129–32.
Hu X, Wu D, Li Y, Wei L, Li X, Qin M, et al. The first familial NSD2 cases with a novel variant in a Chinese father and daughter with atypical WHS facial features and a 7.5-year follow-up of growth hormone therapy. BMC Med Genet. 2020;13(1):181.
Lozier ER, Konovalov FA, Kanivets IV, Pyankov DV, Koshkin PA, Baleva LS, et al. De Novo Nonsense Mutation in WHSC1 (NSD2) in Patient With Intellectual Disability and Dysmorphic Features. J Hum Genet. 2018;63(8):919–22.
Zollino M, Di Stefano C, Zampino G, Mastroiacovo P, Wright TJ, Sorge G, et al. Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet. 2000;94(3):254–61.
Van Buggenhout G, Melotte C, Dutta B, Froyen G, Van Hummelen P, Marynen P, et al. Mild Wolf-Hirschhorn syndrome: micro-array CGH analysis of atypical 4p16.3 deletions enables refinement of the genotype-phenotype map. J Med Genet. 2004;41(9):691–8.
Maas NMC, Van Buggenhout G, Hannes F, Thienpont B, Sanlaville D, Kok K, et al. Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH). J Med Genet. 2008;45(2):71–80.
Yang WX, Pan H, Li L, Wu HR, Wang ST, Bao XH, et al. Analyses of Genotypes and Phenotypes of Ten Chinese Patients with Wolf-Hirschhorn Syndrome by Multiplex Ligation-dependent Probe Amplification and Array Comparative Genomic Hybridization. Chin Med J. 2016;129(6):672–8.
Callaway DA, Campbell IM, Stover SR, Hernandez-Garcia A, Jhangiani SN, Punetha J, et al. Prioritization of Candidate Genes for Congenital Diaphragmatic Hernia in a Critical Region on Chromosome 4p16 using a Machine-Learning Algorithm. J Pediatr Genet. 2018;7(4):164–73.
Bernardini L, Radio FC, Acquaviva F, Gorgone C, Postorivo D, Torres B, et al. Small 4p16.3 deletions: three additional patients and review of the literature. Am J Med Genet A. 2018;176(11):2501–8.
Jiang Y, Sun H, Lin Q, Wang Z, Wang G, Wang J, et al. De novo truncating variant in NSD2gene leading to atypical Wolf-Hirschhorn syndrome phenotype. BMC Med Genet. 2019;20(1):134.
Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, et al. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome. Eur J Hum Genet. 2014;22(4):464–70.
Zollino M, Orteschi D, Ruiter M, Pfundt R, Steindl K, Cafiero C, et al. Unusual 4p16.3 deletions suggest an additional chromosome region for the Wolf-Hirschhorn syndrome–associated seizures disorder. Epilepsia. 2014;55(6):849–57.
Boczek NJ, Lahner CA, Nguyen TM, Ferber MJ, Hasadsri L, Thorland EC, et al. Developmental delay and failure to thrive associated with a loss-of-function variant in WHSC1 (NSD2). Am J Med Genet A. 2018;176(12):2798–802.
Acknowledgments
Not applicable.
Funding
None.
Author information
Authors and Affiliations
Contributions
IB collected the patient’s clinical data, FS performed the genetic analysis, LCW drafted the manuscript, EB edited the manuscript. All authors approved the final version of the manuscript and take full responsibility for its contents.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient’s guardians gave their written consent for publication of this article.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wiel, L.C., Bruno, I., Barbi, E. et al. From Wolf-Hirschhorn syndrome to NSD2 haploinsufficiency: a shifting paradigm through the description of a new case and a review of the literature. Ital J Pediatr 48, 72 (2022). https://doi.org/10.1186/s13052-022-01267-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-022-01267-w