
Abstract
Background
The world’s herbaria contain millions of specimens, collected and named by thousands of researchers, over hundreds of years. However, this treasure has remained largely inaccessible to genetic studies, because of both generally limited success of DNA extraction and the challenges associated with PCR-amplifying highly degraded DNA. In today’s next-generation sequencing world, opportunities and prospects for historical DNA have changed dramatically, as most NGS methods are actually designed for taking short fragmented DNA molecules as templates.
Results
As a practical test of routine recovery of rDNA and plastid genome sequences from herbarium specimens, we sequenced 25 herbarium specimens up to 80 years old from 16 different Angiosperm families. Paired-end reads were generated, yielding successful plastid genome assemblies for 23 species and nuclear rDNAs for 24 species, respectively. These data showed that genome skimming can be used to generate genomic information from herbarium specimens as old as 80 years and using as little as 500 pg of degraded starting DNA.
Conclusions
The routine plastome sequencing from herbarium specimens is feasible and cost-effective (compare with Sanger sequencing or plastome-enrichment approaches), and can be performed with limited sample destruction.
Similar content being viewed by others


Background
Herbaria are collections of preserved plant specimens stored for scientific study. There are approximately 3400 herbaria in the world, containing around 350 million specimens, collected over the past 400 years (http://sciweb.nybg.org/science2/indexHerbariorum.asp). These collections cover most of the world’s plant species, including many rare and endangered local endemics, and species collected from places that are currently expensive or difficult to access [1]. The recovery of DNA from this vast resource of already collected expertly-verified herbarium specimens represent a highly efficient way of building a DNA-based identification resource of the world’s plant species (DNA barcoding) and increasing knowledge of phylogenetic relationships.
The ‘unlocking’ of preserved natural history specimens for DNA barcoding/species discrimination is of particular relevance. In the first decade of DNA barcoding, it became clear that obtaining material from expertly verified is a key rate-limiting step in the construction of a global DNA reference library [2]. The millions of samples that are required for this endeavor, each needing corresponding voucher specimens and meta-data, create a strong impetus for making best-use of previously collected material.
DNA degradation in herbarium samples followed by subsequent diffusion from the sample creates challenges for DNA recovery [3]. In addition, different preservation methods can negatively affect the ability of extract, amplify and sequence DNA [4,5,6]. PCR amplification of historical DNA is, therefore, generally restricted to short amplicons (< 200 bp) and is further vulnerable to contamination by recent DNA and PCR products from the study species. The cumulative damage to the DNA can also cause incorrect bases to be inserted during enzymatic amplification. The main sources for these alterations are single nucleotide misincorporations [7, 8]. Above all, PCR-based Sanger sequencing by using herbarium samples to generate standard DNA barcodes can be challenging. A recent large-scale study by Kuzmina et al. 2017 [9] examined 20,816 specimens representing 5076 of 5190 vascular plant species in Canada. Kuzmina et al. found that specimen age and method of preservation had significant effects on sequence recovery for all barcode markers. However, massively-parallel short-read Next-generation sequencing (NGS) protocols have the potential to greatly increase the success of herbarium sequencing projects, as many new sequencing approaches do not rely on large, intact DNA templates and instead are well-suited for sequencing low concentrations of short (100-400 bp) fragmented molecules [3, 10].
Straub et al. [11], described how “genome skimming”, involving a shallow-pass genome sequence using NGS, could recover highly repetitive genome regions such as rDNA or organelle genomes, and yield highly useful sequence data at relatively low sequence depth, and these regions include the usual suite of DNA barcoding markers [12, 13]. The genome skimming approach using NGS has been used to recover plastid DNA and rDNA sequences from 146 herbarium specimens [14], to produce the entire nuclear genome of a 43-year-old Arabidopsis thaliana herbarium specimen [15], the complete plastome, the mitogenome, nuclear ribosomal DNA clusters, and partial sequences of low-copy genes from an herbarium specimen of an extinct species of Hesperelaea [16, 17], and the complete plastome, nuclear ribosomal DNA clusters, and partial sequences of low-copy genes from three grass herbarium specimens [18].
However, sequencing small, historical specimens may be especially challenging if a specimens is unique, or nearly so, with no alternative specimens available for study should the first specimen fail. Methods used to extract and prepare DNA for sequencing must both be more or less guaranteed to work, and, in many cases, allow for preservation of DNA for future study [19]. In recent studies that report successfully sequencing of historical specimens from 1 ng to 1 μg of input DNA (for example, up to 1 μg in Bakker et al. [14]; ∽ 600 ng in Staats et al. [15]; 33 ng in Zadane et al. [17]; 8.25–537 ng in Kanda et al. [20]; 5.8–200 ng in Blaimer et al. [21]; less than 10 ng in Besnard et al. [18]; 1–10 ng in Sproul and Maddison [19]). But a number of studies also report abandoning a subset of specimens for which too little input DNA was available (i.e. below 10 ng in Kanda et al. [20]; below 5 ng in Blaimer et al. [21]). To better understand ideal approaches of sample preparation for specimens with minimal DNA, we intentionally limited DNA input to 500 pg per specimen.
In this paper we provide a further practical test of the genome skimming methodology applied to herbarium specimens. As part of the China Barcode of Life project, and our wider phylogenomic studies, our aim was to assess whether the success reported in these early genome skimming studies could be repeated in other laboratories.
We evaluated the success and failure rates of rDNA and plastid genome sequencing from genome skims of 25 different species from herbarium specimens, and explored the impacts of parameters such as amount of input DNA and PCR cycle numbers.
Methods
Specimen sampling
25 herbarium specimens were selected from 16 Angiosperm families covering 22 genera, with specimen ages up to 80 years old. All 25 species were taken from the specimens housed in the Herbarium of the Institute of Botany, Chinese Academy of Sciences (KUN). The samples were selected to represent the major clades of APG III system (Table 1).
DNA extraction
Approximately 1 cm2 sections of leaf or 20 mg of leaf tissue were used for each DNA extraction. Genomic DNA was extracted using Tiangen DNAsecure Plant Kit (DP320). Yield and integrity (size distribution) of genomic DNA extracts were quantified by fluorometric quantification on the Qubit (Invitrogen, Carlsbad, California, USA) using the dsDNA HS kit, as well as by visual assessment on a 1% agarose gel.
Library preparation
All samples were subsequently built into blunt-end DNA libraries in the laboratories using the NEBNext Ultra II DNA library Prep kit for Illumina (New England BIolabs) which has been optimized for as little as 5 ng starting DNA and Illumina-specific adapters [22]. The library protocol was performed as per the manufacturer’s instructions with four modifications: (i) 500 pg of input DNA was selected to accommodate low starting DNA quantities, (ii) DNA was not fragmented by sonication because the DNA was highly degraded; (iii) The NEBNext library was generated without any size selection; (iv) DNA libraries were then amplified in an indexing PCR, which barcoded each library and discriminated each sample. Five PCR cycles was suggested by the manufacturer’s instruction for 5 ng of input DNA. As only 500 pg of starting DNA was used, we tested use of increasing numbers of PCR cycles (namely × 6, × 8, × 10, × 12, × 14 PCR cycles). Concentration and size profiles of the final indexed libraries (125 libraries, representing 25 specimens at 5 different numbers of PCR cycles) were assessed on a Bioanalyzer 2100 using a high sensitivity DNA chip.
Library pooling
The final indexed libraries were then pooled (33 or 34 samples per lane) in equimolar ratios and sequenced on three lanes on an Illumina XTen sequencing system (Illumina Inc.) using paired and chemistry at the Cloud health Medical Group Ltd.
Analyses
Successfully sequenced samples were assembled into chloroplast genomes and nuclear rDNAs. Here the rDNAs comprise the complete sequence of 26S, 18S, and 5.8S and internal transcribed spacers (ITS1 and ITS2). We did not assemble the internal gene spacer (IGS) because of the complexity of this region which is rich in duplications and inversions.
The raw sequence reads were filtered for primer/adaptor sequences and low-quality reads with the NGS QC Toolkit [23]. The cut-off value for percentage of read length was 80, and that for PHRED quality score was 30. Then the filtered high-quality pair-end reads were assembled into contigs with Spades 3.0 [24]. Next, we identified highly similar genome sequences using the Basic Local Alignment Search Tool (BLAST: http://blast.ncbi.nlm.gov/). The procedures and parameters for setting the sequence quality control, de novo assembly, and blast search were followed as in Yang et al. [25]. Next, we determined the proper orders of the aligned contigs using the highly similar genome sequences identified in the BLAST search as references. At this point, the target contigs were assembled into complete plastid genomes and nuclear rDNAs.
Annotation of the plastomes was performed using the plastid genome annotation package DOGMA [26] (http://dogma.ccbb.utexas.edu/). Start and stop codons of protein-coding genes, as well as intron/exon positions, were manually adjusted. The online tRNAscan-SE service [27] was used to further determine tRNA genes. The final complete plastomes and rDNAs were deposited into GenBank (Accession numbers: MH394344-MH394431; MH270450-MH270494).
Fungi or other plants may be co-isolated during the DNA extraction process resulting in DNA contamination [1]. This is particularly important where starting DNA concentrations are extremely low. We thus sub-sampled our data to check for contamination. To check for contamination in the plastid DNA sequences, for each species we extracted its rbcL sequence and blasted it against GenBank to check that it grouped with related species. BLAST1 (implemented in the BLAST program, version 2.2.17) was used to search the reference database for each query sequence with an E value < 1 × 10−5. Likewise, to check for plant and fungal contamination in the rDNA sequences, we took the final assembled ITS sequences (or partial ITS sequences where complete ITS was not recovered) and blasted the sequences against the NCBI database to check that it grouped with related species.
Results
All 25 species yielded amounts of DNA suitable for library preparation and further processing. Total yields varied between 3 ng and 400 ng from on average 20 mg of dried leaf tissue, usually the equivalent of 1 cm2 of leaf tissue (Table 1). We found a negative correlation between specimen age and DNA yield (Fig. 1).

DNA yield against specimen age
We successfully enriched and sequenced DNA libraries constructed from herbarium material. Despite only 500 pg of input DNA, good quality libraries were produced from 100 of 125 samples (25 species, with × 8, × 10, × 12, × 14 PCR cycles). The concentration of the final indexed libraries based on six PCR cycles per species was too low to be further sequenced. Between 15,877,478 and 44,724,436 high-quality paired-end reads were produced, with the total number of bases ranging from 2,381,621,700 bp (2.38 giga base pairs, Gbp) to 6,708,665,400 bp (6.71 Gbp) (Table 2). These were then assembled into contigs, and using a blast search into plastid genomes and rDNA arrays.
After de novo assembly, two species (Otochilus porrectus and Pholidota chinensis) generated poor plastid assemblies, with the longest contigs being 6705 bp with 2 × coverage and 1325 bp with 3 × coverage respectively. The other 23 species yielded useful plastid assemblies drawn from 3 to 61 contigs assembled into plastid genomes with depths ranged from 459 × to 2176 ×. Of these 23 species, 14 were assembled into complete plastid genomes. Eight species were assembled into nearly complete plastid genomes, but with gaps ranged from 5 to 349 bp (Table 2). However, although Rhododendron rex subsp. fictolacteum yielded useful plastid assemblies, many gaps were detected among contigs when the species Vaccinium macrocarpon was used as reference data.
For the nuclear rDNAs, 21 species gave ribosomal DNA sequences assemblies > 4.3 kb drawn from 1 to 2 contigs with sequencing depths ranging from 3 × to 567 × (no nrDNA sequences could be assembled for Phodidota chinensis, Paederia scandens, Otochilus porrectus, and Camellia gymnogyna) (Table 3). Of these 21 species, 18 resulted in assembled nrDNAs consisting of partial sequences of 18S and 26S, along with the complete sequence of 5.8S and the internal transcribed spacers ITS1 and ITS2. However, 3 species (2 samples of Manglietia fordiana (Sample ID 01 and 02), Phoebe neurantha (Sample ID 05), were difficult to assemble, resulting in only partial recovery of 5.8S and the internal transcribed spacers ITS1 and ITS2.
To check the quality of the plastid sequences, all gene regions were translated. No stop codons that would be indicative of sequencing errors were detected within the assembled contigs. We then extracted about 1400 bp of rbcL sequence from 23 of the samples to check for contamination (for Rhododendron rex subsp. fictolacteum (Sample ID 16), the plastid genome was not assembled successfully but we could nevertheless extract the rbcL sequence from the plastid contigs). These rbcL sequences were subjected to a blast search against the NCBI database. The rbcL sequences contained no insertions or deletions and matched the correct genus or family in each case (Table 4). Likewise, we blasted the final assembled rDNA ITS sequences (or partial ITS sequences) from 24 samples against the NCBI database. In all cases, the closest match to the sequence was from the family of the sequenced sample. No matches with fungi were detected (Table 5).
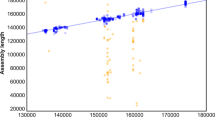
One-way analyses of variance (ANOVA) were performed to test the total reads against PCR cycles, PCR cycles against plastid contig numbers, PCR cycles against plastid genome assembly length, PCR cycles against plastid mean-depth, and PCR cycles against plastid coverage. We found that was no significant correlation between PCR cycles and plastid contig numbers, PCR cycles and plastid genome assembly length, and PCR cycles and plastid coverage. There was, however, a significant positive correlation between the number of PCR cycles and the total number of reads, and PCR cycles and the plastid mean-depth (Fig. 2).

PCR cycles with raw data, contigs, and assembly length
Finally, when comparing plastome assembly coverage with C values of the species concerned we find a slight negative bit not significant correlation (Fig. 3), which would suggest, at least for our sampling, that plastome assembly coverage is not affected by nuclear genome size of the specimen concerned.

Plastome coverage versus C value (pg DNA per 1C) of all samples assembled in this study
Discussion
Sequencing herbarium specimens from low amounts of starting DNA
Our current study successfully demonstrated the recovery of plastid genome sequences and rDNA sequences from herbarium specimens, some up to 80 years old. Our study used small amounts of starting tissue (c 1 cm2) and extremely low initial concentrations (500 pg) of degraded starting DNA. This success with a small amount of starting tissue is important, and demonstrates the practical feasibility of organelle genome and rDNA recovery with minimal impacts on specimens. These findings, in the context of studies by others (e.g. Bakker et al. [14]) confirm that genome skimming can be performed with limited sample destruction enabling relatively straightforward access to high-copy number DNA in preserved herbarium specimens spanning a wide phylogenetic coverage.
To accommodate the use of only 500 pg of input DNA, we modified the library protocol to remove the step of DNA fragmentation by sonication because the DNA was already highly degraded, we did not undertake any size selection, and we increased the number of PCR cycles to enrich the indexed library. After library preparation and Illumina paired-end sequencing, a sufficient number of read pairs (> 15,000,000) were generated for our 25 specimens and 100 libraries. This strategy allowed the generation of complete or near complete plastid genomes with depths ranging from 459 × to 2176 ×, and nuclear ribosomal units with a high sequencing depth (3 × to 567 ×) for 23 and 24 specimens respectively. Despite the low starting concentration, no plant or fungal contaminants were obviously detectable in the assembled plastomes and rDNA sequences.
For herbarium plastome assembly, the procedures and parameters for setting the sequence quality control, de novo assembly, blast search and genome annotation were followed as in Yang et al. [25]. The rate of our 25 specimens with 100 libraries was c. 5 h per specimen on a 3-TB RAM Linux workstation with 32 cores. It was not different significantly between fresh and herbarium specimens.
Recovery of widely used loci in plant molecular systematics
A benefit of the genome skimming approach is that it can recover loci widely used in previous molecular systematics studies (e.g. Coissac et al. 2016 [12]). Here we recovered the standard rbcL DNA barcode region from 23/25 samples, the standard matK DNA barcode region from 23/25 specimens, the standard trnH-psbA DNA barcode region from 23/25 samples, the trnL intron from 23/25 samples, and the ITS1 and ITS2 from 20/25 to 19/25 samples respectively. In addition to the recovery of these standard DNA barcoding loci, we also recovered many other regions used as supplementary barcode markers (e.g. atpF-H, psbK-I). The data produced with this approach can thus contribute towards standard and extended DNA barcode reference libraries [12], in helping identify additional regions which are informative for any given clade [28], as well as producing data for phylogenomic investigations to elucidate the relationships amongst plant groups.
Practical benefits
A primary motivation for this study was our own experiences with suboptimal DNA recovery from herbarium specimens using Sanger sequencing coupled with difficulty in accessing fresh material of some species. The success of this method using only small amounts of starting tissue from herbarium specimens is an important step to addressing these challenges. It makes sequencing type specimens a realistic proposition, which can further serves to integrate genetic data into the existing taxonomic framework. A second practical benefit is that field work is often not possible in some geographical regions where past collections have been made. Political instability and/or general inaccessibility can preclude current collecting activities, and where habitats have been highly degraded or destroyed, the species concerned may simply be no longer available for collection. Mining herbaria to obtain sequences from previously collected material can circumvent this problem. Thirdly, sequencing plastid genomes and rDNA arrays from specimens that are many decades old enables a baseline to be established for haplotype and ribotype diversity. This baseline can then be used to assess evidence for genetic diversity loss or change due to recent population declines or environmental change.
Conclusions
This study confirms the practical and routine application of genome skimming for recovering sequences from plastid genomes and rDNA from small amounts of starting tissue from preserved herbarium specimens. The ongoing development of new sequencing technologies is creating a fundamental shift in the ease of recovery of nucleotide sequences enabling ‘new uses’ for the hundreds of millions of existing herbarium specimens [1, 10, 14, 16, 29]. This shift from Sanger sequencing to NGS approaches has now firmly moved herbarium specimens into the genomic era.
References
Särkinen T, Staats M, Richardson JE, Cowan RS, Bakker FT. How to open the treasure chest? Optimizing DNA extraction from herbarium specimens. PLoS ONE. 2012;7(8):e43808.
Hebert PDN, Hollingsworth PM, Hajibabaei M. From writing to reading the encyclopedia of life. Philos Trans R Soc B. 2016;371(1702):20150321.
Kistler L, Ware R, Smith O, Collins M, Allaby RG. A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. 2017;45(11):6310–20.
Hall LM, Wollcox MS, Jones DS. Association of enzyme inhibition with methods of museum skin preparation. Biotechniques. 1997;22(5):928–34.
Hedmark E, Ellegren H. Microsatellite genotyping of DNA isolated from claws left on tanned carnivore hides. Int J Legal Med. 2005;119(6):370–3.
Tang EPY. Path to effective recovering of DNA from formalin-fixed biological samples in natural history collections: workshop summary. Washington: The National Academies Press; 2006.
Groombridge JJ, Jones CG, Bruford MW, Nichols RA. ‘Ghost’ alleles of the Mauritius kestrel. Nature. 2000;403(6770):616.
Stiller M, Green RE, Ronan M, Simons JF, Du L, He W, Egholm M, Rothberg JM, Keates SG, Ovodov ND, Antipina EE, Baryshnikov GF, Kuzmin YV, Vasilevski AA, Wuenschell GE, Termini J, Hofreiter M, Jaenicke-Després V, Pääbo S. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc Natl Acad Sci USA. 2006;103(37):13578–84.
Kuzmina ML, Braukmann TWA, Fazekas AJ, Graham SW, Dewaard SL, Rodrigues A, Bennett BA, Dickinson TA, Saarela JM, Catling PM, Newmaster SG, Percy DM, Fenneman E, Lauron-Moreau A, Ford B, Gillespie L, Subramanyam R, Whitton J, Jennings L, Metsger D, Warne CP, Brown A, Sears E, Dewaard JR, Zakharov EV, Hebert PDN. Using herbarium-drived DNAs to assemble a large-scale DNA barcode library for the vascular plants of Canada. Appl Plant Sci. 2017;5(12):1700079.
Smith O, Palmer SA, Gutaker R, Allaby RG. An NGS approach to archaeobotanical museum specimens as genetic resources in systematics research. In: Olson PD, Hughes J, Cotton JA, editors. Next generation systematics. Cambridge: Cambridge University Press; 2016. p. 282–304.
Straub SCK, Parks M, Weithmier K, Fishbein M, Cronn RC, Liston A. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. Am J Bot. 2012;99(2):349–64.
Coissac E, Hollingsworth PM, Lavergne S, Taberlet P. From barcodes to genomes: extending the concept of DNA barcoding. Mol Ecol. 2016;25(7):1423–8.
Hollingsworth PM, Li DZ, van der Bank M, Twyford AD. Telling plant species apart with DNA: from barcodes to genomes. Philos Trans R Soc B. 2016;371(1702):20150338.
Bakker FT, Lei D, Yu JY, Mohammadin S, Wei Z, van de Kerke S, Gravendeel B, Nieuwenhuis M, Staats M, Alquezar-Planas DE, Holmer R. Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Biol J Lin Soc. 2016;117(1):33–43.
Staats M, Erkens RHJ, van de Vossenberg B, Wieringa JJ, Kraaijeveld K, Stielow B, Geml J, Richardson JE, Bakker FT. Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS ONE. 2013;8(7):e69189.
Van de Paer C, Hong-Wa C, Jeziorski C, Besnard G. Mitogenomics of Hesperelaea, an extinct genus of Oleaceae. Gene. 2016;594(2):197–202.
Zedane L, Hong-Wa C, Murienne J, Jeziorsky C, Baldwin BG, Besnard G. Museomics Illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Biol J Linnea Soc. 2015;117(1):44–57.
Besnard G, Christin PA, Malé PJG, Lhuillier E, Lauzeral C, Coissac E, Vorontsova MS. From museums to genomics: old herbarium specimens shed light on a C3 to C4 transition. J Exp Bot. 2014;65(22):6711–21.
Sproul JS, Maddison DR. Sequencing historical specimens: successful preparation of small specimens with low amounts of degraded DNA. Mol Ecol Resour. 2017;17:1183–201.
Kanda K, Pflug JM, Sproul JS, Dasenko MA, Maddison DE. Successful recovery of nuclear protein-coding genes from small insects in museums using Illumina sequencing. PLoS ONE. 2015;10:30143929.
Blaimer BB, Lloyd MW, Guillory WX, SnG B. Sequence capture and phylogenetic utility of genomic ultraconserved elements obtained from pinned insect specimens. PLoS ONE. 2016;11:e0161531.
Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010. https://doi.org/10.1101/pdb.prot5448.
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE. 2012;7(2):e30619.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Yang JB, Li DZ, Li HT. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol Ecol Resour. 2014;14(5):1024–31.
Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–5.
Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33(Suppl_2):W686–9.
Li XW, Yang Y, Henry RJ, Rossetto M, Wang YT, Chen SL. Plant DNA barcoding: from gene to genome. Biol Rev. 2015;90(1):157–66.
Hart ML, Forrest LL, Nicholls JA, Kidner CA. Retrieval of hundreds of nuclear loci from herbarium specimens. Taxon. 2016;65(5):1081–92.
Authors’ contributions
BY and DZL organized the project. CXZ performed the experiments, analyzed the data, and wrote the paper; PMH wrote and edited the paper; JY, ZSH, and ZRZ extracted DNA, prepared library. All authors read and approved the final manuscript.
Acknowledgements
We are very grateful to Mr. Wei Fang (Kunming Institute of Botany, Chinese Academy of Sciences) for kindly providing the materials. We would like to thank Ms. Chun-Yan Lin and Mr. Shi-Yu Lv (Kunming Institute of Botany, Chinese Academy of Sciences) for their help with the experiments.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the NCBI SRA repository, SRP142448 and hyperlink to datasets in http://www.ncbi.nlm.nih.gov/home/submit.shtml.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was funded by a program for basic scientific and technological data acquisition of the Ministry of Science of Technology of China (Grant No. 2013FY112600), the Large-scale Scientific Facilities of the Chinese Academy of Sciences (Grant No: 2017-LSF-GBOWS-02), and Biodiversity Conservation Strategy Program of Chinese Academy of Sciences (ZSSD-011).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zeng, CX., Hollingsworth, P.M., Yang, J. et al. Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods 14, 43 (2018). https://doi.org/10.1186/s13007-018-0300-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-018-0300-0