
Abstract
Background
The assessment of TP53 mutational status is becoming a routine clinical practice for chronic lymphocytic leukemia patients (CLL). A broad spectrum of molecular techniques has been employed so far, including both direct Sanger sequencing and next generation sequencing. Oxford Nanopore Technologies recently released the MinION an USB-interfaced sequencer. In this paper we report our experience, with the MinION technology for the detection of the TP53 gene mutation in CLL patients.
Twelve CLL patients at diagnosis were included in this study. All except one patient showed the TP53 gene deletion in Fluorescence in situ hybridization experiments.
Patients were investigated for TP53 mutation by Sanger and by MinION sequencing.
Analysis by Sanger was performed according with the IARC protocol.
Analysis by MinION was performed adopting a strategy based on long template PCR, read error correction, and post variant calling filtering.
Results
Due to the high error rate of nanopore technology, sequence data were both used directly and before correction with two different in silico methods: ALEC and nanocorrect. A mean error rate of 15 % was detected before correction that was reduced to 4-5 % after correction.
Analysis by Sanger sequencing was able to detect four patients mutated for TP53. MinION analysis detected one more mutated patient previously not detected from Sanger.
Conclusion
In our hands, the Nanopore technology shows correlation with Sanger sequencing but more sensitive, manageable and less expensive, and therefore has proven to be a useful tool for TP53 gene mutation detection.
Similar content being viewed by others


Background
Tumor-suppressor p53 gene (TP53) maps to chromosome band 17p13 and is pivotal for genome integrity. TP53 encodes for the p53 protein, a transcription factor involved in essential cell functions, such as DNA repair, cell cycle control, apoptosis, aging, and stemness [1, 2]. Aberrant p53 function, due to 17p deletion (del(17p)) and/or TP53 mutation, is associated with poor prognosis in chronic lymphocytic leukemia (CLL) patients [3–5]. Alterations of TP53 occur in about 10 % of untreated CLL patients [6, 7], but up to 50 % in relapsed or refractory cases [8, 9]. Furthermore, over 80 % of cases harboring del(17p) also carry TP53 mutations in the remaining allele [10, 11]. The frequency of mutations lacking del(17p) varies among different studies depending on the patient cohort and the methodology used, but in general it accounts for about 30 % of all TP53 defects, while sole 17p deletions, without the TP53 mutation, are less frequent, representing about 10 % of all TP53 alterations [12]. Despite TP53 mutation analysis is becoming a routine test for CLL patients, inconsistent results may be obtained among medical centers, possibly due to the variety of methods employed. To reduce the interlaboratory variability, in 2012 the European Research Initiative on CLL (ERIC) published recommendations (recently revised and available at http://www.ericll.org/pages/networks/TP53network) concerning several methodologies suitable for TP53 analysis [13]. Two principal methodological procedures are suggested for TP53 mutation detection: Sanger Sequencing or Next Generation Sequencing (NGS). Generally, at least exons from the fourth to the ninth, including splicing sites, should be covered in the analysis, even if the optimal range goes from the second to the eleventh. According to the ERIC recommendations on sensitivity threshold, only mutations detectable by Sanger sequencing and mutations with an allelic fraction higher than 10 %, if NGS methods are used, should be reported.
NGS is a powerful technology, allowing the detection of many low-rate mutations in potentially every type of disease and sample, but its major limitation remains the high initial investment required for the instrumentation setup. On the other hand, Sanger sequencing is a more affordable method but it is laborious, time consuming, and expensive over time. In this scenario, in 2012, Oxford Nanopore Technologies (ONT) released a portable sequencing device known as MinION [14] and in 2014 launched a community-focused access project: the MinION Access Programme (MAP). MinION is a single-molecule sequencer connected to a laptop through a USB 3.0 interface. Nanopore technology works by connecting two strands of DNA molecules by a hairpin, and sequencing them consecutively. During sequencing, the single strand of DNA passes through biologic nanopores on a chip, where an electric field is applied and electrical signal variations of consecutive 5-mers are recorded. DNA bases are then called using a cloud-based software (Metrichor). Template and complement sequences obtained are then used to generate the 2D high quality sequences. Typically, long reads are produced, up to some dozen kilobases. Due to the still high error rate, at the moment MinION performances cannot be comparable with the previous NGS platforms. However, the very low costs (estimated by the company around USD1000 when it will become commercially available), the ease of use, and the length of the reads, make MinION ideal for screening TP53 mutations, followed by Sanger sequencing validation.
Methods
Patients
Twelve CLL patients at diagnosis were included in this study. All cases showed more than 70 % of lymphocytes in peripheral blood. All except one patient (CLL#7) showed the TP53 gene deletion in Fluorescence in situ hybridization (FISH) experiments, performed as previously reported [15, 16]. The main clinical and biological features of these patients are reported in Table 1. All 12 samples were analyzed by Sanger and MinION in blinded manner.
Sanger sequencing
All samples were analyzed by direct Sanger sequencing according to the International Agency for Research on Cancer (IARC) protocol (http://p53.iarc.fr/Download/TP53_DirectSequencing_IARC.pdf). Electropherograms were then analyzed by visual inspection, glass free software for Sanger analysis data (http://shiny.bat.infspire.org/igcllglass/) and GeneScreen [17].
TP53 Amplification and barcoding
Genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen) and quantified with Qubit 2.0 Fluorometer (Life Technologies).
According to the Oxford Nanopore Barcoding protocol for amplicons (Version DK003_1148_revB_12Feb2015), for each patient, we performed a Long-PCR using the PrimeSTAR GXL DNA Polymearse (Takara Bio Inc.), two tailed primers (PreBAR_P-559 5’-TTTCTGTTGGTGCTGATATTGCTCTCATGCTGGATCCCCACT-3’ and PreBAR_P-E11Re 5’-ACTTGCCTGTCGCTCTATCTTCTGACGCACACCTATTGCAAG-3’), 180 ng of genomic DNA, in a final volume of 25 μl. Thermal-cycling conditions were 98 °C for 10 s, 68 °C for 8 min (30 cycles) and 4 °C hold. The PCR products, visualized on an agarose-gel (1 %), were purified using the QIAquick PCR Purification Kit (Qiagen), and used as templates for the Barcoding PCR. Barcoding was performed with the same Polymerase and 12 different pairs of Barcoding primers from ONT, BC01-BC12, in a final volume of 50 μl. Thermal-cycling conditions were 98 °C for 10 s, 62 °C for 15 s, 68 °C for 8 min (15 cycles) and 4 °C hold.
Minion library preparation and sequencing
Before starting library preparation, we purified, quantified and estimated the purity of samples (Nanodrop). Two mixes were then prepared, each pulling six barcoded amplicons at equal weight ratio. One microgram of each pool was diluted to 80 μl in nuclease-free water and prepared for sequencing. According to the ONT Sequencing protocol (SQK-MAP005), DNA was end-repaired with the NEBNext End Repair Module (New England Biolabs Inc.) and subsequently dA-tailed using the NEBNext dA-Tailing Module (New England Biolabs Inc.), prior to ligation of nanopore-specific adapters with Blunt/TA Ligase Master Mix (New England Biolabs Inc.). All purifications were performed with Agencourt AMPure XP beads (Beckman Coulter Inc.). Dynabeads His-Tag Isolation & Pulldown (Life Technologies) was used to elute the library in the pre-sequencing Mix. After the Platform QC run, sequencing mix (75 μl of 2X Running Buffer, 66 μl of nuclease-free water, 3 μl of Fuel Mix and 6 μl of the Pre-sequencing Mix) was loaded and the MAP_48Hr_Sequencing_Run.py protocol was started (MinION flowcell: FLO-MAP003; MinKNOW software: v48.2.14). During running, two reloads of the sequencing mix were performed and the run was stopped after 24 h.
Data analysis
PoRe R package was used to obtain summary information about the sequencing data, as well as to extract FASTA files from FAST5 files [18].
The NanoOK package was employed for coverage and error assessment, using the TP53 genomic sequence NC_000017.10 as reference [19].
Finally, Galaxy, a web-based platform for processing NGS data, was employed for variant analysis (https://usegalaxy.org/), [20–22]. Reads were aligned on GRCh37 human reference genome with the BWA-MEM method [23] using specific Nanopore platform parameters (https://github.com/lh3/bwa/blob/master/NEWS.md/#release-079-19-may-2014) and visualized with the Integrative Genomics Viewer (IGV) browser [24, 25]. Single nucleotide variants (SNV) and insertions/deletions (indels) detection was separately performed with the Varscan software [26], and VCF files were filtered and annotated with the gene variation IARC TP53 database (v. R17). In particular, for SNV detection, the minimum read depth and supporting reads parameters were set according to the lowest mean coverage (minimum read depth of 50, minimum supporting reads of 10). Whereas, for indels detection, the minimum read depth and the minimum supporting reads values were set at 50 and 13, respectively. The minimum variant allele frequency threshold and p-value threshold for variant calling were 0.01 and 0.05, respectively, in all Varscan analyses.
Results
TP53 mutation analysis by Sanger sequencing
All patients included in our study were investigated for TP53 mutation by Sanger sequencing, according with the IARC protocol.
In the attempt of increasing our analytical sensitivity, we decided to test the MinION performance on selected CLL cases bearing the 17p deletion. As assessed by FISH, 11/12 patients carried the 17p deletion, and among them, 4 also had a TP53 mutation (Tables 1 and 3). Reportedly [10], there is a high chance to find TP53 mutations in patients with the 17p deletion, thus supporting our decision in case selection.
TP53 mutation analysis by MinION sequencing
TP53 gene was amplified from genomic DNA in a single Long PCR reaction. Amplicons, spanning from exon 2 to exon 11 (7150 bp), were then barcoded and loaded on MinION for sequencing.
A total of two runs were performed, employing two libraries of six patients each one and two different flowcells, with 316 and 401 active pores respectively. For both experiments, 22421 fast5 total files were produced, containing raw electric signals. After base-calling in Metrichor, each run generated 14Mbases and 24Mbases. Finally, of the total reads produced, 3619 passed 2D filters and 2652 had a recognizable barcode (Table 2). In general, 2D pass reads had a length spanning from 150 bp to 7200 bp with a mean of around 4 Kb. Plotting the distribution of read lengths, we could observe a prevalent peak around 7 Kb, in agreement with our amplicon size (Fig. 1). Shorter reads, likely originating from breaks during purifications or library preparation, were observed as well.

Reads length distribution plot
Data analysis and error correction
Nanopore sequencing is a relative young technology and despite the rapid technical improvements, is still affected by a high error rate (estimated around 10 %), posing a problem in SNV analysis. To date, the only data on human gene variant analysis from MinION sequencing regard the detection of structural variants in cancer [27].
Among the several methods proposed to limit the error rate, we tested two in silico strategies: the Nanocorrect correction pipeline, specific for Nanopore data [28], and the more generic ALEC (Amplicon Long-read Error Correction) python script [29].
Nanocorrect is a pipeline inspired by pbdagcon (https://github.com/PacificBiosciences/pbdagcon) which uses DALIGNER (https://github.com/thegenemyers/DALIGNER) and poa [30] to correct sequencing errors in Nanopore reads. ALEC script has been originally developed for processing PacBio raw data to correct random sequencing errors specifically on long amplicons using alignment information from the SAM files [29].
After demultiplexing, reads were analyzed before and after error correction with NanoOK (Additional file 1: Table S1). A mean error rate of 15 % on mean values per 100 aligned bases counted including indels was detected before correction (raw reads), whereas the mean error rate dropped to 4-5 % after correction with nanocorrect and ALEC. Furthermore, separate error rate analysis for insertions, deletions and substitutions highlighted higher values for deletions. Mean insertion/deletion size reported was 1-2 nucleotides.
Coverage analyses showed that the TP53 sequence, from exon 2 to exon 11, introns included, was completely and uniformly covered in each patient (Fig. 2). Moreover, as compared to Nanocorrect corrected reads, raw data and ALEC corrected data generally produced higher coverage rates, although the minimum coverage value was never below 50x.

Coverage plots from corrected and uncorrected (Raw) reads after mapping on TP53 genomic sequence
Variant calling and annotation
Read mapping, variant calling, annotation with “gene variation IARC TP53 database”, and variant filtering were performed with Galaxy starting from both raw reads and corrected reads. We created a complete workflow available TP53 at https://usegalaxy.org/u/ematlab/w/tp53-mutation-screening-filtering to use with a reformatted file of “gene variation IARC TP53 database” (Additional file 2: File S2). After analysis the workflow outputs two files for SNV and Indels already filtered against TP53 mutation IARC database.
Variant filtering
Excluding intronic non-affecting splicing and exonic silent variations, analyses with raw reads returned the highest number of variants, especially for SNV. Conversely, the most stringent output was obtained from variant calling performed on nanocorrect corrected reads. Results were further filtered by Sanger sequencing detection limit, set around 15 % for SNV and 20 % for indels due to the higher error rate observed for deletions (Additional file 1: Table S1).
To evaluate whether recurrent sequencing errors could occur because of the nature of the sequences, thus producing false positives results, we checked if same variants were called in different patients. We found some recurrent variants both in samples sequenced on the same flowcell and on different flowcells, and therefore we decided also to filter variants by recurrence, considering the low chance to find the same mutation in more than one sample simultaneously.
Comparing the results obtained from corrected and uncorrected reads after filtering, we found 24 unique variants called from uncorrected reads, and 17 and 8 called from ALEC and nanocorrect corrected reads respectively.
A final summary of mutations detected with all methods we used and their comparison is shown in Table 3.
In general, all mutations detected by Sanger sequencing were also detected starting from raw reads and ALEC corrected reads. Additionally, for patient CLL#12, one mutation previously not detected by automated electropherogram analysis, was instead detected by visual inspection after the indications generated from MinION (Fig. 3, Table 3 and Additional file 3: Fig S3). Nanocorrect corrected reads produced two false negatives, namely in CLL#8 and CLL#12 (Table 3).

Example of mutations detected in two samples (CLL#8 and CLL#12). The mutation detected in CLL#8 was not found from nanocorrect corrected reads. CLL#12 mutation was detected only from the analysis with raw and ALEC corrected reads. The same mutation was barely visible in Sanger sequencing. Aligned reads are visualized by IGV
As expected, all recurrent variants resulted as false positive.
Discussion
The MinION measures changes in electrical current as individual strands of DNA pass through one of its 500 protein tiny pores. To date, it is the only technology that directly measures a single DNA strand rather than incorporation events relative to a template strand. Moreover, speed, single-base sensitivity, and long read lengths make nanopores a promising technology for high-throughput sequencing [14, 31, 32]. In particular, the ability to generate long reads may help to resolve repeat regions within the genome that are challenging for short-read platforms; in fact, it has been recently demonstrated that MinION is able to determine the number of repeats in the human X chromosome repeat region using spanning reads of 42 Kbp [33]. This feature makes nanopore-based technology potentially very suitable for the targeted sequencing of cancer-relevant gene mutations. The only limitation of MinION technology is error proneness, an issue that can be moderated by several error correction bioinformatics methods.
In the present work we report the first use of MinION to detect TP53 mutations in CLL patients. The recommended method to assess TP53 mutation status is standard Sanger sequencing; pre-screening techniques such as denaturing high-performance liquid chromatography (dHPLC) or single-strand conformation analysis polymorphism (SSCP) could be performed to reduce time and costs. In any case results have to be confirmed by Sanger sequencing in order to identify the nature of mutation.
TP53 mutation analysis in CLL patients is particularly challenging considering that TP53 aberrations are infrequent at the time of initial chemotherapy, but increase their prevalence in multiply relapsed, chemotherapy-refractory populations [6, 8]. Therefore, monitoring TP53 status after the first line treatment or at CLL relapse is of considerable importance in order to plan the best treatment option for the patient.
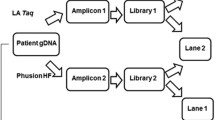
Using our version of MinION (MIN-MAP001) we developed a workflow (Fig. 4) for read error correction together with a variant calling pipeline that considerably reduce the false positive variants, thus improving the overall performance. In our hands, the nanopore-based technology has proven to be a useful tool for TP53 mutation detection. From a practical point of view, in the context of TP53 mutational analysis, we suggest that MinION sequencing could be ideal in a pre-screening step, with several advantages compared to the current methods. Additionally, MinION pre-screening approach requires only two PCR per patient (TP53 long amplification and barcoding) instead of one PCR per exon. The obtained results are more informative in comparison to those obtained with other pre-screening methods, potentially making the subsequent Sanger interpretation easier and more sensitive. Anyway at the state of the art of this technology, from a clinically point of view we recommend to comply at the ERIC reporting guidelines for Sanger sequencing (http://www.ericll.org/pages/networks/tp53network/ericmanualfortp53mutationalanalysis).

Schematic workflow implemented for TP53 mutational analysis
As mentioned, one of the major drawbacks is the high error rate that might lead to an elevated number of false positive variants. To limit this error, we tested two different in silico methods for reads correction: nanocorrect and ALEC, and applied additional filters on the variants called, such as IARC annotation, mutation effect, allelic frequency and recurrence. Mainly, recurrence was the most effective filter to reduce false positives rate, but required multiple samples to be performed.
As regards error correction methods, nanocorrect strongly reduced the final variants called, although produced some false negatives. Conversely, raw reads did not return false negative but too many false positives were called. The best results were obtained by ALEC, which returned no false negatives and a very limited number of false positives.
Finally, a relevant advantage of MinION is its limited costs. Excluding device charge, that is estimated around USD1000, we calculated that, if 6 samples are contemporarily run in a flowcell, the cost per sample is around USD180 a price comparable to that of Sanger sequencing. Moreover, since very high coverages were reached in our analyses, we believe that the overall costs could be further reduced by running more samples per flowcell.
Conclusions
In summary, we demonstrate that MinION is a suitable tool for the detection of TP53 gene mutations in patients affected by CLL, and propose that its low costs and ease of use may potentially expand its field of applications to other cancer genes. At the moment MinION is still not adequate for substituting Sanger sequencing nor other NGS technologies, but can be a useful strategy for pre-screening analyses. Finally, the constant improvements of nanopore technology promise an exclusive and convenient use of MinION in the immediate future.
Abbreviations
- ALEC:
-
Amplicon long-read error correction
- BC:
-
Barcode
- bp:
-
Base pair
- BWA-MEM:
-
Burrows-wheeler aligner – maximal exact match
- CLL:
-
Chronic lymphocytic leukemia
- del(17p):
-
17p deletion
- dHPLC:
-
Denaturing high-performance liquid chromatography
- DNA:
-
Deoxyribonucleic acid
- dNTP:
-
deoxynucleotide triphosphate
- ERIC:
-
European Research Initiative on CLL
- FISH:
-
Fluorescence in situ hybridization
- IARC:
-
International Agency for Research on Cancer
- IGV:
-
Integrative Genomics Viewer
- Indel:
-
Insertion/deletion
- Kb:
-
Kilobase pair
- MAP:
-
MinION access program
- NGS:
-
Next-generation sequencing
- ONT:
-
Oxford nanopore technologies
- PCR:
-
Polymerase chain reaction
- SNV:
-
Single nucleotide variant
- SSCP:
-
Single-strand conformation analysis polymorphism
- TP53:
-
Tumor protein p53
- VCF:
-
Variant call format
References
Lu W-J, Amatruda JF, Abrams JM. p53 ancestry: gazing through an evolutionary lens. Nat Rev Cancer. 2009;9:758–62.
Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–9.
Dreger P, Corradini P, Kimby E, Michallet M, Milligan D, Schetelig J, et al. Indications for allogeneic stem cell transplantation in chronic lymphocytic leukemia: the EBMT transplant consensus. Leukemia. 2007;21:12–7.
Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N Engl J Med. 2000;343:1910–6.
Mauro FR, Molica S, Laurenti L, Cortelezzi A, Carella AM, Zaja F, et al. Fludarabine plus alemtuzumab (FA) front-line treatment in young patients with chronic lymphocytic leukemia (CLL) and an adverse biologic profile. Leuk Res. 2014;38:198–203.
Zenz T, Krober A, Scherer K, Habe S, Buhler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112:3322–9.
Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, Van Dyke DL, O’Brien S, et al. De novo deletion 17p13.1 chronic lymphocytic leukemia shows significant clinical heterogeneity: the M. D. Anderson and Mayo Clinic experience. Blood. 2009;114:957–64.
Tam CS, O’Brien S, Lerner S, Khouri I, Ferrajoli A, Faderl S, et al. The natural history of fludarabine-refractory chronic lymphocytic leukemia patients who fail alemtuzumab or have bulky lymphadenopathy. Leuk Lymphoma. 2007;48:1931–9.
Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114:2589–97.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29:2223–9.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15:995–1004.
Malcikova J, Pavlova S, Kozubik KS, Pospisilova S. TP53 mutation analysis in clinical practice: lessons from chronic lymphocytic leukemia. Hum Mutat. 2014;35:663–71.
Pospisilova S, Gonzalez D, Malcikova J, Trbusek M, Rossi D, Kater AP, et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia. 2012;26:1458–61.
Loman NJ, Quinlan AR. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics. 2014;30:3399–401.
Storlazzi CT, Albano F, Locunsolo C, Lonoce A, Funes S, Guastadisegni MC, et al. t(3;12)(q26;q14) in polycythemia vera is associated with upregulation of the HMGA2 gene. Leukemia. 2006;20:2190-2.
Albano F, Anelli L, Zagaria A, Coccaro N, Minervini A, Rossi AR, et al. Decreased TET2 gene expression during chronic myeloid leukemia progression. Leuk Res. 2011;35:e220-2.
Carr IM, Camm N, Taylor GR, Charlton R, Ellard S, Sheridan EG, et al. GeneScreen: a program for high-throughput mutation detection in DNA sequence electropherograms. J Med Genet. 2011;48:123–30.
Watson M, Thomson M, Risse J, Talbot R, Santoyo-Lopez J, Gharbi K, et al. poRe: an R package for the visualization and analysis of nanopore sequencing data. Bioinformatics. 2014;31:114–5.
Leggett RM, Heavens D, Caccamo M, Clark MD, Davey RP. NanoOK: multi-reference alignment analysis of nanopore sequencing data, quality and error profiles. Bioinformatics. 2015;32:142–4.
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, et al. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;Chapter 19:Unit 19.10.1–21.
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–5.
Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76.
Norris AL, Workman RE, Fan Y, Eshleman JR, Timp W. Nanopore sequencing detects structural variants in cancer. Cancer Biol Ther. 2016;17:246–53.
Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12:733–5.
Qiao W, Yang Y, Sebra R, Mendiratta G, Gaedigk A, Desnick RJ, et al. Long-Read Single Molecule Real-Time Full Gene Sequencing of Cytochrome P450-2D6. Hum Mutat. 2016;37:315–23.
Lee C, Grasso C, Sharlow MF. Multiple sequence alignment using partial order graphs. Bioinformatics. 2002;18:452–64.
Quick J, Quinlan AR, Loman NJ. A reference bacterial genome dataset generated on the MinION(TM) portable single-molecule nanopore sequencer. Gigascience. 2014;3:22.
Rusk N. Genomics: MinION takes center stage. Nat Methods. 2014;12:12–3.
Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12:351–6.
Acknowledgements
The authors would like to thank Ms. MVC Pragnell, B.A. for language revision of the manuscript.
Funding
This work was supported by “Associazione Italiana contro le Leucemie (AIL) - BARI”
Availability of data and material
The sequence data from this study have been submitted to the NCBI Short Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession n° SRP080948.
Authors’ contributions
CFM and FA conceived and designed the study, and wrote the manuscript. CC and CFM performed the Long-PCRs, Barcoding and nanopore experiments. PO and CFM performed all bioinformatics analysis. PC performed conventional cytogenetic analysis. LA, AZ, NC and GT conducted FISH experiments and interpreted data. AM, LI and CB performed diagnostic molecular analysis. AG provided clinical data. GS and FA supervised the manuscript preparation. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved (n. 0953) by the ethics committee of the Azienda Ospedaliero-Universitaria Consorziale Policlinico di Bari (Bari, Italy). The written informed consent was obtained from the patients included in this study.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Error analysis results on raw and corrected reads. (XLSX 11 kb)
Additional file 2:
Reformatted file of “gene variation IARC TP53 database” to use in Galaxy workflow for variants annotation. (TXT 1603 kb)
Additional file 3: Figure S3.
Sanger analysis on CLL#12 showing the mutation g.7578406C > T. It is not detectable by the analysis software due to the low intensity of variant peak. The presence of the peaks for T base both in forward and in reverse sequences confirm the mutation. (TIF 3259 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Minervini, C.F., Cumbo, C., Orsini, P. et al. TP53 gene mutation analysis in chronic lymphocytic leukemia by nanopore MinION sequencing. Diagn Pathol 11, 96 (2016). https://doi.org/10.1186/s13000-016-0550-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-016-0550-y