
Abstract
Background
As rabies still represents a major public threat with tens of thousands of deaths per year, particularly in developing countries, adequate surveillance based on rapid and reliable rabies diagnosis for both humans and animals is essential. Rabies diagnosis relies on highly sensitive and specific laboratory tests for detection of viral antigens. Among those tests, at present the immunofluorescence antibody test is the “gold standard test” for rabies diagnosis, followed by virus isolation in either mice or cell culture. Because of the advantages of molecular assays in terms of sensitivity and applicability their approval as confirmatory diagnostic test by international organizations (OIE, WHO) is envisaged. Therefore, the objective was to develop and validate novel molecular assays and RNA extraction methods for rabies that reduce the turnaround time but remain highly sensitive and specific.
Methods
Here, novel assays, i.e. HighSpeed RT-qPCR and isothermal recombinase polymerase amplification (RPA) were designed and tested. Furthermore, three magnetic bead-based rapid extraction methods for manual or automated extraction were validated and combined with the new downstream assays.
Results
While the conventional column based RNA extraction method showed the highest intra-run variations, all magnetic bead-based rapid extraction methods delivered nearly comparable sensitivity and efficiency of RNA recovery. All newly developed molecular tests were able to detect different rabies virus strains in a markedly reduced timeframe in comparison to the standard diagnostic assays. The observed detection limit for the HighSpeed RT-qPCR was 10 genome copies per reaction, and 1000 genome copies per reaction for the RPA assay.
Conclusion
Magnetic bead-based rapid RNA extraction methods are highly sensitive and show a high level of reproducibility and therefore, are particularly suitable for molecular diagnostic assays including rabies. In addition, with a detection limit of 10 genome copies per reaction, the HighSpeed RT-qPCR is suitable for rapid ante mortem rabies diagnosis in humans as well as confirmatory test in integrated bite management and subsequent post-exposure prophylaxis.
Similar content being viewed by others


Background
Rabies is a lethal zoonotic disease caused by a group of 16 negative-strand RNA viruses of the genus Lyssavirus in the family Rhabdoviridae of the order Mononegavirales [1]. It is a societal tragedy that in the twenty-first century, rabies, a zoonosis that can easily be prevented in humans and controlled in domestic animal species is still neglected and continues to create a significant social and economic burden on a global scale [2,3,4]. In low-income countries where control efforts are lacking and awareness of the disease and access to appropriate preventive and post–exposure prophylaxis is limited or non–existent, rabies is estimated to still cause around 60,000 human deaths per year [5, 6].
The real impact of this neglected zoonotic disease is likely to be underestimated. One major problem here is surveillance, which unfortunately is inadequate or even non-existing in many Asian and African countries, where the burden of rabies is highest [6,7,8,9]. Adequate rabies surveillance in both humans and animals goes hand in hand with rapid and reliable rabies diagnosis. Currently, rabies diagnosis relies on laboratory tests for detection of viral antigens. Among those tests, the fluorescence antibody test (FAT) is the “gold standard test” in post mortem rabies diagnosis [6, 10]. Recently, alternative antigen detection methods such as ELISA, the direct rapid immunohistochemical test (DRIT) or the indirect rapid immunohistochemistry test (IRIT) have been developed which do not require expensive fluorescence microscopic equipment along with the expertise and financial input needed to maintain them [11,12,13,14,15]. The rabies tissue culture infection test (RTCIT) or the mouse inoculation test (MIT) are mainly used as confirmatory tests and based on the complex and time consuming propagation and isolation of the virus [16, 17]. All these test methods, however, are (i) dependent on the quality of the supplied sample, or (ii) require longer turnaround times [6, 18]. Easy to use and swift test systems for rapid, cost-efficient diagnosis, with no loss of sensitivity or specificity would therefore improve the diagnostic situation significantly [12, 18, 19]. In this respect, immunochromatographic strip tests, also called lateral-flow devices (LFDs) or antigen-capture point-of-care tests, have a great potential [20,21,22,23,24,25,26], however for rabies there is still a need for standardization and quality check before being an alternative for rapid and simple diagnostics in resource-limited settings [27].
In the course of the past three decades, molecular tools based on the detection of the genetic information of rabies virus have become more widely accepted for the diagnosis of rabies [18]. The development of reverse transcription polymerase chain reaction (RT-PCR) techniques provided an alternative method for post mortem rabies diagnosis [28, 29], and enabled rapid ante mortem diagnosis of human rabies [30,31,32]. Next to conventional or real-time RT-PCRs for rabies [18, 32,33,34,35,36,37], alternative rapid genome detection tests have been developed, i.e. nucleic acid sequence based amplification and reverse-transcriptase loop-mediated isothermal amplification (RT-LAMP) [34, 38,39,40,41,42,43]. Other nucleic acid based methods, i.e. HighSpeed RT-qPCR and isothermal recombinase polymerase amplification (RPA), successfully used for detection of other viral pathogens including Schmallenberg virus (SBV), bovine viral diarrhea virus (BVDV), or foot-and-mouth disease virus (FMDV) [44,45,46] have not been applied for RABV yet. Also, initial efforts to optimize and shorten the process of nucleic acid extraction [47] by remaining highly sensitive and specific have not been further explored.
Therefore, the aim of this study was to (i) design and test both a HighSpeed RT-qPCR and a RT-RPA assay for rapid detection of the RABV, (ii) test the suitability of novel RNA extraction methods that improve reproducibility, analytical sensitivity and operational performance for brain tissue and (iii) combine them with the new downstream assays using different amplification kits. Those tests could then be considered both in advanced as well as in standard laboratories for the detection and confirmation of rabies virus, and thus contribute to the development of better tests for rapid, economical diagnosis for rabies.
Methods
Samples
Lyssavirus positive (FAT positive) brain material was provided by the WHO Collaborating Centre for Rabies Surveillance and Research (FLI, Isle of Riems, Germany) comprising field samples and samples from animal experiments (N = 12) including the sample subset II (N = 43, Table 4) described in a previous study for the evaluation of commercial LFDs for rabies [27]. RABV negative brain tissues (N = 8) were obtained from cattle, wolves, sheep and bats.
Extraction methods
In this study, three magnetic bead-based RNA extraction methods were compared in terms of efficiency to the widely accepted centrifuge-based TRIzol (Life technologies, Darmstadt, Germany) & RNeasy (QIAGEN, Hilden, Germany) method. For each extraction 20 mg of brain tissue was applied.
-
i)
Manual extraction using TRIzol Reagent + RNeasy mini kit
After homogenization of the brain samples with a steel bead in 1 ml TRIzol Reagent (Life technologies, Darmstadt, Germany), chloroform was added and RNA from the aqueous phase was precipitated with 75% ethanol. RNA was further purified with RNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions and eluted in 50 μl RNase free water.
-
ii)
Manual extraction using the SpeedXtract Virus kit
The brain samples were homogenized with a steel bead in 1 ml PBS and nucleic acids were extracted by the SpeedXtract Virus Kit (QIAGEN, Hilden, Germany) using an adapted protocol with an initial enrichment step. Briefly, 200 μl brain homogenate, 400 μl enrichment Buffer EN (QIAGEN) and 30 μl SpeedXtract Suspension A (magnetic beads) were mixed and incubated. After separation of the magnetic beads in a magnetic stand (Invitrogen), the beads have been washed with 500 μl Buffer EN. Then, the magnetic beads were re-suspended in 200 μl FCPL buffer-master-mixture and lysed by heat (95 °C for 5 min). Finally, 100 μl of the nucleic acid containing supernatant were transferred to a new microtube.
-
iii)
Automated extraction using the EZ1 robotic system
The EZ1 Advanced XL (QIAGEN) is a fully automated nucleic acid extraction. Here, the extraction protocol optimized by Aebischer et al. (2014) [47] was applied. In summary, the DNA Blood Card (QIAGEN) was used, as it represents the shortest extraction protocol (processing time 16 min). For sample lysis, 200 μl homogenized brain sample was mixed 1:1 with buffer VXL (all reagents from QIAGEN). The elution volume was set to 100 μl.
-
iv)
Automated extraction using the KingFisher Duo
Extraction with the KingFisher (KF) Duo platform (Thermo Fisher Scientific, Waltham, MA, USA) was optimized for rapid extraction of blood and serum by Aebischer et al. (2014) [47]. The published protocol has a processing time of only 8 min. The buffers used were identical with the ones of the EZ1 extraction approach but were handled in the 96 deep-well plate format of the KF Duo. The extracted RNA was eluted in 100 μl.
Reproducibility of extraction methods
To compare the reproducibility of the standard TRIzol & RNeasy method with the other rapid methods, a single RABV positive brain sample (FLI laboratory ID 21867) was extracted in four biological replicates in three independent runs. All extracted RNAs were analyzed with the standard R14 RT-qPCR [34].
Linearity and analytic sensitivity of extraction methods
A ten-fold dilution series of a RABV-positive brain (21867) was prepared. For each extraction method, the samples were tested in duplicates and analyzed with the standard R14 RT-qPCR assay as described below. PCR efficiencies were calculated based on the resulting standard curves.
Analytical performance and operational analysis of extraction methods
For analytical performance, eleven RABV-positive archived brain samples from one dog (21,867 (five dilutions)), one horse (1913) and five foxes (1919, 3690, 3695, 14,067, 17,040), as well as one RABV-negative brain were extracted with the four different extraction methods. RABV-specific RNA was then detected with the R14 RT-qPCR assay and the internal control measured in parallel. In order to compare the extraction methods concerning time-to-extract (operational analysis), twelve samples were extracted in parallel and the respective time was measured.
Amplification and detection methods
For validation of the amplification and detection tests all RNAs were extracted using the manual TRIzol & RNeasy method.
-
i)
Standard RT-qPCR
For amplification and detection of the RABV genome, the standard RT-qPCR assay “R14” was applied [34]. Extraction success was further evaluated using a heterologous HEX-labeled internal control system [48]. The specific RT-qPCR assay was performed using the One-step RT-PCR AgPath Kit (Thermo Fisher Scientific, Waltham, USA). The reaction mix was compromised of 2.25 μl H2O, 6.25 μl 2× RT-PCR buffer, 0.5 μl 25× RT-PCR enzyme mix, 0.5 μl primer-probe mix for the internal control and 0.5 μl R14 primer-probe mix. As template 2.5 μl extracted RNA were used. The temperature profile was 10 min reverse transcription at 45 °C, 10 min activation at 95 °C followed by 42 cycles of 15 s 95 °C, 20 s 56 °C and 30 s 72 °C [35]. A BioRad CFX96 Real-time detection system (version 3.1) was used resulting in a run-time of 95 min.
-
ii)
HighSpeed RT-qPCR
The HighSpeed RT-qPCR was developed on the R14-FAM assay, using the same primers and probes [34]. The SensiFAST Probe No-Rox One-step Kit (Bioline, Luckenwalde, Germany) was used with 2.0 μl H2O, 5.0 μl 2× SensiFAST Probe Mix, 0.2 μl reverse transcriptase, 0.3 μl RNase inhibitor and 0.5 μl R14-Mix-FAM per reaction, resulting in concentrations of 500 nM RV-N-196-F, 500 nM RV-N-283-R, 125 nM RabGT1-B-FAM in 10 μl of mastermix, which was mixed with 2 μl of extracted RNA. The final temperature profile for the BioRad CFX96 Real-time detection system (version 3.1) was 5 min at 45 °C for reverse transcription followed by 10 s at 95 °C for activation. Then 40 cycles of 1 s at 95 °C for denaturation, and 2 s at 60 °C for annealing, extension and measuring were done. Total run-time was 31 min.
-
iii)
RT-RPA
Based on a RABV MAFFT [49] alignment of all representative full-length sequences of the species rabies lyssavirus available in GenBank, primers and probes suitable for RT-RPA were designed and tested. For amplification the RT-RPA chemistry TwistAmp Exo RT Kit (TwistDx, Cambridge, United Kingdom) was used. The mastermix (22.75 μl) contained 420 nM forward primer RABV-N-71Fv4 (5’ATG GAT GCC GAC AAG ATT GTM TTY AAA GTY AAT AAT CA 3′), 420 nM reverse primer RABV-N-211Rv1 (5′ ATG GAT GCC GAC AAG ATT GTM TTY AAA GTY AAT A 3′) and 120 nM probe RABV-N-196-antisense (5′ TCA AAT CTT TGA TAG CAG GGT ACT TGT ACT CA(FAM-dT) AT(THF) GA(BHQ-dT) CCA CGA TAA TC 3′; FAM-dT = deoxythymidine nucleoside derivated with the fluorophore FAM; THF = tetrahydrofuran; BHQ-dT = deoxythymidine nucleoside carrying a blackhole quencher 1), 14.75 μl rehydration buffer and 5.6 μl RNase free water. The volume for two reactions was applied to rehydrate a single lyophilized reagent pellet, as this contains enzymes and chemicals for a final reaction volume of 50 μl. Afterwards, the rehydrated reaction mix was split into two halves, and the general volume of the reaction was reduced to 25 μl. Finally, 22.75 μl mastermix were mixed with 1 μl of extracted RNA. To start the reaction, 1.25 μl of magnesium acetate (MgOAc) were added. The isothermal reaction was incubated for 15 min at 40 °C in an ESEQuant TS2 instrument (QIAGEN Lake Constance, Stockach, Germany), just interrupted by vortexing the amplification reactions after 5 min. The instrument measured the fluorescence signal in 30 s intervals. Real-time fluorescence data were analyzed using a threshold-based method (ESEQuant TS2 studio software version 1.9.3).
Linearity and analytic sensitivity of detection methods
A ten-fold dilution series of extracted and quantified RNA from three different RABV strains (SAD B19, Kelev and Bobcat) was tested with the three different detection methods to determine the analytical sensitivity.
Diagnostic sensitivity and specificity of novel molecular detection methods
In order to determine the suitability of the detection methods, a test panel of RNA from field samples (sample subset II [27]) was evaluated in duplicates with the standard method as well as the two rapid detection methods.
Combination of different extraction methods with novel downstream assays
Eight brain homogenates (one dog (21867), one horse (1913), five foxes (1919, 3690, 3695, 14,067, 17,040) and one negative cow), extracted with four different extraction methods, were tested with three different RABV detection methods to test the suitability of the input material for combined assays for routine diagnostics. Furthermore, the overall analysis time rounded in minutes from sample-to-result was measured.
Statistics
Regression analysis and the Pearson’s correlation coefficient were calculated to compare the extraction and detection methods. Graphics were made using SigmaPlot v11. PCR efficiencies were calculated using the formula E = 10(−1/slope)-1 with the slopes of the standard curves.
Results
Rapid RNA extraction methods
Previous unpublished proof of principle tests showed that both published magnetic bead-based automated extraction methods (EZ1 and KF Duo) as well as the new SpeedXtract (SXT) manual extraction kit were all suitable for nucleic acid extraction out of homogenized brain material.
Reproducibility of RNA extraction methods
Mean Cq values as well as intra- and inter-run variabilities for the different extraction methods are shown in Table 1. The lowest reproducibility was detected with the standard manual extraction method (TRIzol & RNeasy), whereas the best results were obtained with the manual extraction method SXT. Both extraction instruments delivered comparable results.
Linearity and analytical sensitivity of RNA extraction methods
Analytical sensitivity of the tested extraction methods was determined using a ten-fold dilution series of RABV-positive brain. The manual standard TRIzol & RNeasy extraction method and the EZ1 extraction detected both RABV RNA up to a 10−4 dilution, whereas the manual SXT and the KF Duo were more sensitive and detected even the 10−6 dilution step. Linearity was good for all four methods indicated by correlation coefficients of 0.98 for the automated extractions and 0.99 for the manual extractions, respectively (Fig. 1). PCR efficiencies were calculated from standard curves of the dilution series. The lowest efficiency (92%) was observed for the standard procedure, followed by the EZ1 method (97%). The KF Duo (113%) and the SXT (118%) yielded comparably higher efficiencies.

Linearity and analytical sensitivity of the four different extraction methods. A RABV-positive brain homogenate was diluted ten-fold. Numbers indicate mean values for each method. Extracted RNA was quantified with the R14 standard RT-qPCR. R2 values are indicated for each method
Analytical performance of RNA extraction methods
A panel of field samples was extracted to assess the diagnostic sensitivity of the rapid extraction protocols for RABV. The Cq values of the standard TRIzol & RNeasy detection method were compared with the Cq values resulting from the other three methods in a Bland-Altman plot (Fig. 2). The other manual extraction method, SXT, had the lowest correlation to the standard procedure with a coefficient of 0.97. The average Cq value difference was −1.03 (SD = 0.52). Automated EZ1 extraction showed a correlation coefficient of 0.98 and a mean Cq value difference of +0.97 (SD = 0.45), therefore representing a weaker sensitivity. KF Duo also delivered a correlation coefficient for the extractions of 0.98 with a mean Cq value difference of −0.49 (SD = 0.78) (Fig. 2). Furthermore, the heterologous internal control, IC2, was measured in the HEX channel and delivered similar results for three extraction methods, only the EZ1 method showed higher Cq values. The measured mean Cq value with TRIzol & RNeasy was 28.29, for SXT 28.33, for EZ1 30.31 and for KF Duo 28.34.

Analytical performance of rapid extraction methods using RABV samples. Ten RABV field samples were extracted using the standard TRIzol & RNeasy method as well as with a) SpeedXtract, b) EZ1 and c) KF Duo. Cq values of the R14 RT-qPCR as well as internal controls were compared
Operational analysis of RNA extraction methods
The time required to extract twelve samples with the four different methods is given in Table 2. The most time-consuming method is the standard manual extraction procedure, which takes about 45 min for the total extraction process including 35 min hands-on time. In contrast, the other three methods require considerably less hands-on time (less than 25 min). The second longest extraction method is the EZ1 procedure taking 41 min. This includes 25 min hands-on time with 15 min thereof dedicated to filling reagent into the cartridge. The KF Duo method takes 24 min in total including 16 min hands-on time. The fastest method was SXT with only 20 min in total and 15 min hands-on time.
Rapid amplification and detection using HighSpeed RT-qPCR and RT-RPA assay
Like the standard R14 RT-qPCR assay (amplicon length 87 nt) the two novel rapid detection methods target the RABV N-gene. For the HighSpeed RT-qPCR three different kits (SensiFAST Probe No-Rox One-step Kit (Bioline, Luckenwalde, Germany), Ag-Path ID One-Step RT-PCR kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA), qScript XLT 1-Step RT-qPCR ToughMix (QuantaBio, Beverly, Massachusetts, USA)) were tested. Furthermore, modified temperature profiles were used on three different thermocyclers (BioRad CFX96 Real-time detection system, Illumina Eco Thermal Cycler and Roche LightCycler Nano) to optimize and shorten the existing R14 RT-qPCR. For the RT-RPA the best primer and TwistAmp Exo probe were chosen (amplicon length 140 bp), which led to a reduction of the incubation time. Only the final protocols are given in the material and method section. For better comparability of the results, all Cq values were converted into detection times as RT-RPA is measured in minutes. The total run time of the different amplification methods was 95 min for normal RT-qPCR, 31 min for HighSpeed RT-qPCR, and 15 min for RT-RPA being the fastest method.
Linearity and analytical sensitivity of detection methods
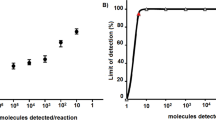
The three different amplification and detection methods vary in their sensitivity. The standard R14 RT-qPCR was the most sensitive method. All three RABV strains could be amplified down to one target RNA copy per μl reaction with a good linearity shown by a correlation coefficient of 0.99 for all strains. The HighSpeed RT-qPCR assay was slightly less sensitive with a detection limit of 10 target RNA copies per μl reaction for the strains SAD B19 and Bobcat USA. Only the Kelev strain could be detected with 1 target RNA copy per μl reaction. Also for the HighSpeed RT-qPCR assay an R2 value of 0.99 for all strains could be ascertained. RT-RPA amplification yielded the least analytical sensitivity. With this method the determined detection limit was 1000 target RNA copies per μl reaction for all three RABV strains. For the Kelev strain no correlation coefficient could be set, because only two dilution steps were detected. For SAD B19 the R2 was 0.94 and for Bobcat USA the R2 was 1.0, respectively.
Please note that the starting concentrations of the viral load for SAD B19 and Bobcat USA were 105 copies/μl reaction, whereas Kelev started with 104 copies/μl reaction. A comparison of the detection time of each method reveals a distinct difference between rapid assay (less than 30 min) and standard RT-qPCR (longer than 40 min) (Fig. 3).

Linearity and analytical sensitivity of the three detection methods. Ten-fold dilution series of three extracted and quantified RABV strains were tested with three different detection methods. Regression lines are illustrated and the correlation coefficient given in the legend
Diagnostic sensitivity and specificity of detection methods
All three assays detected a broad range of different RABV samples (N = 33). The standard RT-qPCR and the HighSpeed amplification method showed a diagnostic sensitivity of 100% (Table 3). The only method showing one false-negative result was the RT-RPA assay, which missed to detect a RABV sample from North America (diagnostic sensitivity 97.0%). The dilution series of RABV sample 20,299 was amplified and detected by all methods. The other tested lyssaviruses showed only one cross detection. DUVV was detected with all three methods with relatively late detection times, whereas EBLV-1, EBLV-2 and BBLV delivered no false-positive results. No amplification of negative samples could be observed.
Combination of different extraction methods with downstream assays
Our analysis showed that the final detection results of specific nucleic acid extracts is independent of the application of the downstream amplification assay. The standard R14 assay detected all positive samples between 40 and 60 min. The two rapid assays returned results in less than 20 min (Fig. 4). Consequently, an assessment of the total processing time from sample to result for each combination of methods revealed notably large differences. The routine diagnostic procedure combining TRIzol & RNeasy with R14 RT-qPCR takes more than two hours, whereas SpeedXtract together with RT-RPA takes only about 35 min (Fig. 5).

Diagnostic results of eight samples analyzed with R14 RT-qPCR, HighSpeed RT-qPCR and RT-RPA. The previous extraction was made with a) manual TRIzol & RNeasy, b) manual SpeedXtract, c) automated EZ1 and d) automated KF Duo. Total time until detection is given in minutes

Turnaround time from sample to result for all combinations of nucleic acid extraction methods with amplification and detection methods
In addition, the four extraction methods (Table 4) and the three detection methods (Table 5) were compared concerning their suitability as a POCT, for example regarding costs, stability of reagents, robustness, speed and other aspects. Thereby SXT and RT-RPA performed the best.
Discussion
Although new techniques have been developed for the diagnosis of rabies in recent years, the number of laboratory confirmed human rabies cases from the most affected countries is limited and represents an underestimate of the real impact of the disease [19, 29]. Therefore, the WHO stresses the need for better tests for rapid and economical diagnosis, with no loss of sensitivity or specificity [6]. The quick and simple to perform diagnostic tests would be ideal for use particularly in countries where laboratory infrastructure is still unfavorable [18, 20]. In the range of diagnostic assays molecular tests are getting more and more attention [18, 29]. Because of the higher sensitivity and specificity over antigen directed assays the molecular nucleic acid amplification tests, i.e. PCRs, are increasingly used as comparative standard detection methods [18, 32,33,34,35,36,37], although they are not recommended for routine use by international organizations yet [6]. Interestingly, proficiency trials among European laboratories showed that PCRs had less false negative results compared to virus isolation using the standard RTCIT [17].
In this study, we developed and validated specific and sensitive rapid molecular detection methods for the detection of RABV, compared to a published assay and improved the performance of the assays by the combination with novel RNA extraction methods. The results show that simplification of this kind of assays with no relevant loss of sensitivity or specificity can be achieved; thus encouraging both their (i) acceptance as a rapid confirmatory test to first-line assays and (ii) extension of its application in many laboratories, thus improving the overall diagnostic capacities.
However, for detection and amplification of lyssaviral target genomes high quality extraction of RNA is crucial. Manual RNA extractions are prone to errors and relatively time-consuming, but also conventional automated extraction techniques often have lengthy procedures and need trained staff. Recently, automated rapid magnetic bead-based RNA extraction methods have been described but only optimized for blood and serum samples [47]. Therefore, three commercial rapid extraction methods were tested regarding their suitability for brain tissue as the material of choice for post-mortem rabies diagnosis and compared to standard extraction method. In terms of reproducibility, the TRIzol & RNeasy method showed the highest intra-run variations (6.6%), and the three rapid extraction methods delivered nearly comparable results. While in the latter case, the EZ1 method as a fully-automated platform was assumed to show lowest coefficients of variation, interestingly best results were obtained with the manual SpeedXtract (SXT) method (2%) (Table 1). Although variations due to variable sample input or samples taken from a different brain region cannot be completely ruled out, sensitivity and efficiency of RNA recovery was acceptable for all rapid RNA extraction methods, with highest rates for SXT as well as KF Duo (Fig. 1). All three tested rapid RNA extraction methods used magnetic particles for nucleic acid extraction but request different instrument equipment. The advantage of the reverse extraction procedure of SXT is that it only requires a heat block and a magnetic stand, whereas the other two rapid methods employ technically sophisticated instrumentation. Both instruments differ enormously in size and weight: the KF Duo is much smaller (40 cm × 46 cm × 34 cm; 17 kg) than the EZ1 (51 cm × 57 cm × 57 cm; 48 kg) making it more attractive for use in smaller laboratories or integration into a mobile laboratory. Regarding the processing time, again SXT and KF Duo performed best (Table 2). The longer total hands-on time for the EZ1 is due to the extensive pre-filling step of cartridges, hence, commercially available cartridges are not suitable for application with shortened extraction protocol. It must be emphasized, however, that the EZ1 method developed by Aebischer et al. includes a self-assembly step, where empty EZ1 cartridges are filled with optimized extraction buffers [47]. Consequently, this EZ1-based method currently is not commercially available and its use depends on the supply of eligible kits (Table 4).
Reduced turnaround times and improved applicability of molecular detection methods can be achieved in different ways. Usually, HighSpeed RT-PCRs take advantage of special but expensive PCR machines with fast heating and cooling ramps, but even with a standard thermocycler (CFX96) an enormous reduction of reaction time for an established detection protocol (R14 RT-qPCR to HighSpeed RT-qPCR) can be achieved.
While the use of thermocyclers depend on a permanent power supply, isothermal amplification methods like RPA can be run, for example, on the ESEQuant TS2; a portable stand-alone, battery powered instrument with multi-channel real-time fluorescence detection capabilities. Highly specific loop-mediated isothermal amplification (LAMP), rolling circle amplification (RCA), strand-displacement amplification (SDA) or helicase dependent amplification (HDA) are either relatively cumbersome in terms of primers design, not rapid enough or costly despite relatively easy primer design and fast amplification [44]. RPA is based on the formation of a recombinase filament, strand displacement and abasic nucleotide analogons and has a broad temperature spectrum [50]. Even successful amplification of target RNA by RPA using body heat has been shown [51]. A further advantage of the RPA method is that in contrast to RT-qPCRs the dried reagents of RT-RPA do not require a cold chain. Unfortunately, the lower analytical sensitivity of RT-RPA (1000 target RNA copies per μl reaction) with a smaller dynamic range seems to run the advantages futile (Fig. 3). This analytic sensitivity seems to be comparable to other described isothermal amplification methods for rabies [39, 42]. However, these methods are proposed as suitable for rabies diagnostics. Although the tested RNAs represent different lyssavirus species and major RABV lineages from different regions of the world, the genetic diversity is still not fully covered and the methods requires further validation. However, at least for the HighSpeed RT-qPCR, which is basically a high speed version of the standard R14 RT-qPCR [34] a combination as multiplex or parallel assays with the R13 RT-qPCR, a slightly modified version of the Wakeley protocol [52], could help to overcome the diversity among RABVs and limit the chance of false negatives tremendously [34].
Despite lower analytical sensitivity, RPA in combination with SpeedXtract would be a promising candidate for simplified molecular diagnosis of rabies. Furthermore, molecular detection methods seem to be more reliable than the diagnostic standard FAT in decomposed animals [53]. As the viral load in brain tissue of rabid animals is relatively high, the methods can aid and enhance the passive laboratory confirmed surveillance under resource-limited settings [27]. Furthermore, the simple test approaches with no requirement for cold chain, could even be integrated into mobile test systems, as shown for food-and-mouth-disease virus or Ebola virus [45, 54] (Fig. 5; Tables 4 and 5).
Conclusions
Commercial magnetic bead-based rapid RNA extraction methods are suitable for brain tissue and show high sensitivity as well as a high level of reproducibility compared to standard RNA extraction methods and thus, can help standardizing RNA extraction and molecular diagnostics under routine conditions.
Furthermore, the here presented proof-of-principle simplified rapid rabies virus molecular detection methods (HighSpeed RT-qPCR and RPA) showed almost no loss of diagnostic sensitivity or specificity compared to validated standard molecular assays. Thus, the simple, quick and sensitive virus RNA extraction from brain samples combined with fit-for–purpose detection methods may improving the reliability and acceptance for the rapid molecular diagnosis of rabies. Particularly the high analytical sensitivity makes the HighSpeed RT-qPCR a potential candidate as a method of choice as rapid (i) ante mortem diagnostics for human rabies, (ii) differential diagnostics for organ transplantations and (iii) confirmatory diagnostics to first-line assays including integrated bite management and subsequent post-exposure prophylaxis. Especially under resource-limited settings, the SpeedXtract based RNA extraction combined with the RPA detection of the rabies genome could be a useful and robust molecular diagnostic approach.
Change history
14 November 2017
After publication of the article [1], it has been brought to our attention that an incorrect genetic sequence has been cited. On page 4, paragraph 1 the following sequence is cited “ATG GAT GCC GAC AAG ATT GTM TTY AAA GTY AAT A 3”. This is an error and the correct sequence should be “GGG GGC TTT YCC TAG GGT KAT ACW GGG CTT T 3”.
Abbreviations
- FAT:
-
Fluorescent antibody test
- LAMP:
-
Loop-mediated isothermal amplification
- LFD:
-
Lateral flow device
- PCR:
-
Polymerase chain reaction
- PEP:
-
Post-exposure prophylaxis
- qPCR:
-
Quantitative/real-time polymerase chain reaction
- RABV:
-
Rabies virus
- RPA:
-
Recombinase polymerase amplification
- RT:
-
Reverse transcription
References
Afonso CL, Amarasinghe GK, Banyai K, Bao Y, Basler CF, Bavari S, Bejerman N, Blasdell KR, Briand FX, Briese T, et al. Taxonomy of the order Mononegavirales: update 2016. Arch Virol. 2016;181:2351–60.
Fooks AR: Rabies remains a 'neglected disease'. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin 2005, 10(11):211–212.
Wunner WH, Briggs DJ. Rabies in the 21 century. PLoS Negl Trop Dis. 2010;4(3):e591.
Rupprecht CE, Turmelle A, Kuzmin IV. A perspective on lyssavirus emergence and perpetuation. Current opinion in virology. 2011;1(6):662–70.
Rupprecht CE, Blass L, Smith K, Orciari LA, Niezgoda M, Whitfield SG, Gibbons RV, Guerra M, Hanlon CA. Human infection due to recombinant vaccinia-rabies glycoprotein virus. N Engl J Med. 2001;345(8):582–6.
World Health Organisation. Expert Consultation on Rabies, Second report. World Health Organ Tech Rep Ser. 2013;982:1–150.
Knobel DL, Cleaveland S, Coleman PG, Fevre EM, Meltzer MI, Miranda ME, Shaw A, Zinsstag J, Meslin FX. Re-evaluating the burden of rabies in Africa and Asia. Bull World Health Organ. 2005;83(5):360–8.
Baxter JM. One in a million, or one in thousand: What is the morbidity of rabies in India? Journal of global health. 2012;2(1):010303.
Hampson K, Coudeville L, Lembo T, Sambo M, Kieffer A, Attlan M, Barrat J, Blanton JD, Briggs DJ, Cleaveland S, et al. Estimating the global burden of endemic canine rabies. PLoS Negl Trop Dis. 2015;9(4):e0003709.
Dean DJ, Abelseth MK. Laboratory techniques in rabies: the fluorescent antibody test. Monograph series World Health Organization. 1973;23:73–84.
Bourhy H, Rollin PE, Vincent J, Sureau P. Comparative field evaluation of the fluorescent-antibody test, virus isolation from tissue culture, and enzyme immunodiagnosis for rapid laboratory diagnosis of rabies. J Clin Microbiol. 1989;27(3):519–23.
Lembo T, Niezgoda M, Velasco-Villa A, Cleaveland S, Ernest E, Rupprecht CE. Evaluation of a direct, rapid immunohistochemical test for rabies diagnosis. Emerg Infect Dis. 2006;12(2):310–3.
Xu G, Weber P, Hu Q, Xue H, Audry L, Li C, Wu J, Bourhy H. A simple sandwich ELISA (WELYSSA) for the detection of lyssavirus nucleocapsid in rabies suspected specimens using mouse monoclonal antibodies. Biologicals. 2007;35(4):297–302.
Madhusudana SN, Subha S, Thankappan U, Ashwin YB. Evaluation of a direct rapid immunohistochemical test (dRIT) for rapid diagnosis of rabies in animals and humans. Virol Sin. 2012;27(5):299–302.
Dyer JL, Niezgoda M, Orciari LA, Yager PA, Ellison JA, Rupprecht CE. Evaluation of an indirect rapid immunohistochemistry test for the differentiation of rabies virus variants. J Virol Methods. 2013;190(1–2):29–33.
Webster WA, Casey GA: Virus isolation in neuroblastoma cell culture. In: Laboratory techniques in rabies. Edited by Meslin FX, Kaplan MM, Koprowski H, 4th edn. Geneva: World Health Organization; 1996: 93–104.
Robardet E, Picard-Meyer E, Andrieu S, Servat A, Cliquet F. International interlaboratory trials on rabies diagnosis: An overview of results and variation in reference diagnosis techniques (fluorescent antibody test, rabies tissue culture infection test, mouse inoculation test) and molecular biology techniques. J Virol Methods. 2011;177(1):15–25.
Fooks AR, Johnson N, Freuling CM, Wakeley PR, Banyard AC, McElhinney LM, Marston DA, Dastjerdi A, Wright E, Weiss RA, et al. Emerging technologies for the detection of rabies virus: challenges and hopes in the 21st century. PLoS Negl Trop Dis. 2009;3(9):e530.
Dürr S, Naissengar S, Mindekem R, Diguimbye C, Niezgoda M, Kuzmin I, Rupprecht CE, Zinsstag J. Rabies diagnosis for developing countries. PLoS Negl Trop Dis. 2008;2(3):e206.
Kang B, Oh J, Lee C, Park BK, Park Y, Hong K, Lee K, Cho B, Song D. Evaluation of a rapid immunodiagnostic test kit for rabies virus. Journa of Virological Methods. 2007;145(1):30–6.
Nishizono A, Khawplod P, Ahmed K, Goto K, Shiota S, Mifune K, Yasui T, Takayama K, Kobayashi Y, Mannen K, et al. A simple and rapid immunochromatographic test kit for rabies diagnosis. Microbiol Immunol. 2008;52(4):243–9.
Markotter W, York D, Sabeta CT, Shumba W, Zulu G, Le Roux K, Nel LH. Evaluation of a rapid immunodiagnostic test kit for detection of African lyssaviruses from brain material. The Onderstepoort journal of veterinary research. 2009;76(2):257–62.
Servat A, Picard-Meyer E, Robardet E, Muzniece Z, Must K, Cliquet F. Evaluation of a Rapid Immunochromatographic Diagnostic Test for the detection of rabies from brain material of European mammals. Biologicals : journal of the International Association of Biological Standardization. 2012;40(1):61–6.
Voehl KM, Saturday GA. Evaluation of a rapid immunodiagnostic rabies field surveillance test on samples collected from military operations in Africa, Europe, and the Middle East. US Army Medical Department journal. 2014:27–32.
Lechenne M, Naissengar K, Lepelletier A, Alfaroukh IO, Bourhy H, Zinsstag J, Dacheux L. Validation of a Rapid Rabies Diagnostic Tool for Field Surveillance in Developing Countries. PLoS Negl Trop Dis. 2016;10(10):e0005010.
O’Farrell B: Evolution in Lateral Flow–Based Immunoassay Systems. Springer 2009, In: Wong RC, Tse HY, editors:1–33.
Eggerbauer E, de Benedictis P, Hoffmann B, Mettenleiter TC, Schlottau K, Ngoepe EC, Sabeta CT, Freuling CM, Muller T. Evaluation of Six Commercially Available Rapid Immunochromatographic Tests for the Diagnosis of Rabies in Brain Material. PLoS Negl Trop Dis. 2016;10(6):e0004776.
Sacramento D, Bourhy H, Tordo N. PCR technique as an alternative method for diagnosis and molecular epidemiology of rabies virus. Mol Cell Probes. 1991;5(3):229–40.
Dacheux L, Wacharapluesadee S, Hemachudha T, Meslin FX, Buchy P, Reynes JM, Bourhy H. More accurate insight into the incidence of human rabies in developing countries through validated laboratory techniques. PLoS Negl Trop Dis. 2010;4(11):e765.
Crepin P, Audry L, Rotivel Y, Gacoin A, Caroff C, Bourhy H. Intravitam diagnosis of human rabies by PCR using saliva and cerebrospinal fluid. J Clin Microbiol. 1998;36(4):1117–21.
Panning M, Baumgarte S, Pfefferle S, Maier T, Martens A, Drosten C. Comparative analysis of rabies virus reverse transcription-PCR and virus isolation using samples from a patient infected with rabies virus. J Clin Microbiol. 2010;48(8):2960–2.
Mani RS, Madhusudana SN, Mahadevan A, Reddy V, Belludi AY, Shankar SK. Utility of real-time Taqman PCR for antemortem and postmortem diagnosis of human rabies. J Med Virol. 2014;86(10):1804–12.
Coertse J, Weyer J, Nel LH, Markotter W. Improved PCR methods for detection of African rabies and rabies-related lyssaviruses. J Clin Microbiol. 2010;48(11):3949–55.
Hoffmann B, Freuling CM, Wakeley PR, Rasmussen TB, Leech S, Fooks AR, Beer M, Muller T. Improved safety for molecular diagnosis of classical rabies viruses by use of a TaqMan real-time reverse transcription-PCR "double check" strategy. J Clin Microbiol. 2010;48(11):3970–8.
Fischer M, Freuling CM, Muller T, Wegelt A, Kooi EA, Rasmussen TB, Voller K, Marston DA, Fooks AR, Beer M, et al. Molecular double-check strategy for the identification and characterization of European Lyssaviruses. J Virol Methods. 2014;203:23–32.
Faye M, Dacheux L, Weidmann M, Diop SA, Loucoubar C, Bourhy H, Sall AA, Faye O. Development and validation of sensitive real-time RT-PCR assay for broad detection of rabies virus. J Virol Methods. 2017;
Wadhwa A, Wilkins K, Gao J, Condori Condori RE, Gigante CM, Zhao H, Ma X, Ellison JA, Greenberg L, Velasco-Villa A, et al. A Pan-Lyssavirus Taqman Real-Time RT-PCR Assay for the Detection of Highly Variable Rabies virus and Other Lyssaviruses. PLoS Negl Trop Dis. 2017;11(1):e0005258.
Wacharapluesadee S, Hemachudha T. Nucleic-acid sequence based amplification in the rapid diagnosis of rabies. Lancet. 2001;358(9285):892–3.
Boldbaatar B, Inoue S, Sugiura N, Noguchi A, Orbina JR, Demetria C, Miranda ME, Yamada A. Rapid detection of rabies virus by reverse transcription loop-mediated isothermal amplification. Jpn J Infect Dis. 2009;62(3):187–91.
Saitou Y, Kobayashi Y, Hirano S, Mochizuki N, Itou T, Ito FH, Sakai T. A method for simultaneous detection and identification of Brazilian dog- and vampire bat-related rabies virus by reverse transcription loop-mediated isothermal amplification assay. J Virol Methods. 2010;168(1–2):13–7.
Hayman DT, Banyard AC, Wakeley PR, Harkess G, Marston D, Wood JL, Cunningham AA, Fooks AR. A universal real-time assay for the detection of Lyssaviruses. J Virol Methods. 2011;177(1):87–93.
Wacharapluesadee S, Phumesin P, Supavonwong P, Khawplod P, Intarut N, Hemachudha T. Comparative detection of rabies RNA by NASBA, real-time PCR and conventional PCR. J Virol Methods. 2011;175(2):278–82.
Muleya W, Namangala B, Mweene A, Zulu L, Fandamu P, Banda D, Kimura T, Sawa H, Ishii A. Molecular epidemiology and a loop-mediated isothermal amplification method for diagnosis of infection with rabies virus in Zambia. Virus Res. 2012;163(1):160–8.
Aebischer A, Wernike K, Hoffmann B, Beer M. Rapid genome detection of Schmallenberg virus and bovine viral diarrhea virus by use of isothermal amplification methods and high-speed real-time reverse transcriptase PCR. J Clin Microbiol. 2014;52(6):1883–92.
Abd El Wahed A, El-Deeb A, El-Tholoth M, Abd El Kader H, Ahmed A, Hassan S, Hoffmann B, Haas B, Shalaby MA, Hufert FT, et al. A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PloS one. 2013;8(8):e71642.
Euler M, Wang Y, Heidenreich D, Patel P, Strohmeier O, Hakenberg S, Niedrig M, Hufert FT, Weidmann M. Development of a panel of recombinase polymerase amplification assays for detection of biothreat agents. J Clin Microbiol. 2013;51(4):1110–7.
Aebischer A, Beer M, Hoffmann B. Development and validation of rapid magnetic particle based extraction protocols. Virol J. 2014;11:137.
Hoffmann B, Depner K, Schirrmeier H, Beer M. A universal heterologous internal control system for duplex real-time RT-PCR assays used in a detection system for pestiviruses. J Virol Methods. 2006;136(1–2):200–9.
Katoh K, Misawa K, Kuma K-I, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66.
Piepenburg O, Williams CH, Stemple DL, Armes NA. DNA detection using recombination proteins. PLoS Biol. 2006;4(7):e204.
Crannell ZA, Rohrman B, Richards-Kortum R. Equipment-free incubation of recombinase polymerase amplification reactions using body heat. PLoS One. 2014;9(11):e112146.
Nadin-Davis SA, Sheen M, Wandeler AI. Development of real-time reverse transcriptase polymerase chain reaction methods for human rabies diagnosis. J Med Virol. 2009;81(8):1484–97.
McElhinney LM, Marston DA, Brookes SM, Fooks AR. Effects of carcase decomposition on rabies virus infectivity and detection. J Virol Methods. 2014;207:110–3.
Faye O, Faye O, Soropogui B, Patel P, El Wahed AA, Loucoubar C, Fall G, Kiory D, Magassouba N, Keita S et al: Development and deployment of a rapid recombinase polymerase amplification Ebola virus detection assay in Guinea in 2015. Euro surveillance : bulletin Europeen sur les maladies transmissibles = European communicable disease bulletin 2015, 20(44).
Acknowledgements
We thank Christian Korthase and Jeannette Kliemt at the Friedrich-Loeffler-Institute for excellent technical assistance. Further, we thank Merle Hanke, Claudia Engemann, Andy Wende, and Christian Fischer-Rasokat at QIAGEN for technical support and for providing SpeedXtract kits.
Funding
This work was supported by the FLI intern funded research network “Lyssavirus – a potential public health risk”.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: KS BH CMF TM. Performed the experiments: KS. Analyzed the data: KS BH. Wrote the paper: KS, BH, CMF, TM, MB. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
None. All samples were collected in the frame of routine rabies surveillance or were available from previous animals trials.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
A correction to this article is available online at https://doi.org/10.1186/s12985-017-0894-2.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schlottau, K., Freuling, C.M., Müller, T. et al. Development of molecular confirmation tools for swift and easy rabies diagnostics. Virol J 14, 184 (2017). https://doi.org/10.1186/s12985-017-0853-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-017-0853-y