
Abstract
Background
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant muscular disorder characterized by asymmetric muscle wasting and weakness. FSHD can be subdivided into two types: FSHD1, caused by contraction of the D4Z4 repeat on chromosome 4q35, and FSHD2, caused by mild contraction of the D4Z4 repeat plus aberrant hypomethylation mediated by genetic variants in SMCHD1, DNMT3B, or LRIF1. Genetic diagnosis of FSHD is challenging because of the complex procedures required.
Methods
We applied Nanopore CRISPR/Cas9-targeted resequencing for the diagnosis of FSHD by simultaneous detection of D4Z4 repeat length and methylation status at nucleotide level in genetically-confirmed and suspected patients.
Results
We found significant hypomethylation of contracted 4q-D4Z4 repeats in FSHD1, and both 4q- and 10q-D4Z4 repeats in FSHD2. We also found that the hypomethylation in the contracted D4Z4 in FSHD1 is moderately correlated with patient phenotypes.
Conclusions
Our method contributes to the development for the diagnosis of FSHD using Nanopore long-read sequencing. This finding might give insight into the mechanisms by which repeat contraction causes disease pathogenesis.
Similar content being viewed by others
Background
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal disease characterized by muscle weakness that initially manifests in the face, shoulder, and upper arms, followed by asymmetric involvement of other muscles [1]. DUX4 is a causative gene for FSHD and is located within an approximately 3.3 kb repeat sequence, referred to as D4Z4, which comprises 1–100 repeat units (RUs) on the subtelomeric regions of chromosomes 4 and 10. Chromosome 4 has two haplotypes distal of the D4Z4 repeat, 4qA and 4qB, where only the 4qA allele contributes to FSHD development, due to the presence of a polyadenylation signal in the most distal D4Z4 RU [2, 3].
FSHD has two types, FSHD1 and FSHD2, both caused by genetic defects leading to aberrant DUX4 expression in skeletal muscle [4]. FSHD1 is mediated by contraction of the D4Z4 4qA allele to 1–10 RUs [5], while FSHD2 is caused by a combination of milder D4Z4 contraction (8–20 RUs) and genetic variants in SMCHD1, DNMT3B, or LRIF1, which each encode epigenetic modifiers [6,7,8]. Epigenetic modifiers affect histone modification, DNA methylation, and RNA-based mechanisms, may be involved in mechanisms of various diseases and have important diagnostic potential [9] DNA methylation and histone modification at D4Z4 RUs are altered in FSHD [10,11,12]. CpG methylation is specifically decreased at the contracted D4Z4 repeat on chromosome 4 in FSHD1, while the D4Z4 repeats on both chromosomes 4 and 10 are hypomethylated in FSHD2 [10, 13, 14]; however, the distribution of methylation throughout the full D4Z4 repeat sequence has not been analyzed.
Southern blotting, bisulfite sequencing, molecular combing, and next-generation sequencing are currently used for genetic diagnosis of FSHD [15], but these diagnostic procedures and interpretation of their results present several difficulties. First, interpretation of hybridization patterns generated by Southern blotting is complicated by the fact that the detecting probe also recognizes an additional locus on chromosome 10q that is almost completely homologous to the target 4q35 locus. Second, two subtelomeric variations distal to D4Z4 have been identified on chromosome 4, referred to as the 4qA and 4qB alleles, and selective identification of contracted 4qA repeats is necessary, as only 4qA is associated with FSHD. Third, analysis of CpG methylation by bisulfite sequencing has been performed across the entire D4Z4 units at both the 4q and 10q loci; however, a focal region of extreme demethylation has been reported [16]. Additionally, several patients with milder D4Z4 contraction and CpG hypomethylation have been identified, making diagnosis difficult.
Here, we applied Nanopore CRISPR/Cas9-targeted resequencing (nCATS) to measure the number of D4Z4 RUs and their methylation status in patients with FSHD. We specifically analyzed D4Z4 RUs derived from 4qA and measured the CpG methylation rate in each RU. D4Z4 RUs from 10q were also analyzed.
Methods
Genomic DNA preparation
Peripheral blood lymphocytes (10 ml) were combined with 30 ml EL buffer (155 mM NH4Cl, 10 mM KHCO3, 1 mM EDTA, pH 7.4) on ice for 15 min, followed by centrifugation (KUBOTA 5930, RS-3012M) (840×g, 10 min, room temperature). After a repeat EL buffer wash, pellets were suspended in 3 ml NL buffer (10 mM Tris–HCl, 2 mM EDTA, 400 mM NaCl, pH 8.2), followed by addition of 1% SDS and proteinase K and incubation at 37 °C overnight. DNA lysis solution was added with 1 ml 5 M NaCl, followed by phenol/chloroform extraction and ethanol precipitation. DNA pellets were suspended in TE buffer.
Fibroblasts grown in culture dishes were lysed in 10 mM Tris–HCl, 10 mM EDTA, 150 mM NaCl, pH 8.0 containing 0.5% SDS and proteinase K at 55 °C overnight, followed by phenol/chloroform extraction and ethanol precipitation. DNA pellets were suspended in TE buffer.
DNA library preparation
DNA libraries were prepared using a ligation sequencing kit (Oxford Nanopore Technologies, SQK-LSK109). To generate Cas9 ribonucleoprotein complexes (RNPs), annealed 1 μM tracrRNA-crRNA pool (CR1/CR2/CR3/CR4) and 0.5 μM HiFi Cas9 were incubated at room temperature (around 23 °C) for 30 min. Genomic DNA (2 μg) was dephosphorylated with Quick Calf Intestinal Phosphatase (NEB, #M0525S) at 37 °C for 10 min, followed by 80 °C for 2 min. For Cas9 RNP cleavage and dA-tailing, dephosphorylated genomic DNA samples were treated with Cas9 RNPs, Taq polymerase (NEB, #M0273S), and dATP (NEB, #N0440S) at 37 °C for 30 min, followed by 72 °C for 5 min. For native barcode ligation, native barcoding expansion 1–12 (Oxford Nanopore Technologies, EXP-NBD104) were ligated to cleaved and dA-tailed genomic DNAs using Blunt/TA Ligase Master Mix (NEB, #M0367L) at room temperature for 10 min, followed by purification with Agencourt AMPure XP Beads (Beckman Coulter, #A63880) on a magnet. AMII adapters were ligated to barcoded genomic DNA using Quick T4 DNA ligase (NEB, #E7185A) at room temperature for 10 min, followed by purification with AMPure XP Beads on a magnet. The DNA library from Cas9-targeted native barcoding was primed into a MinION Flow Cell (FLO-MIN106D) on a MinION Mk1C and sequencing was performed for 20–21 h.
The crRNA design tool, CHOPCHOP [17], was used to design crRNAs, which were synthesized by Integrated DNA Technologies as follows: CR1, 5′gataccgacagcaatagtcc3′; CR2, 5′gtccttcagcactccacatc3′; CR3, 5′ctataggatccacagggagg3′; and CR4, 5′tgtcaaggtttggcttatag3′.
Data analysis
Bases were called from Fast5 files using Guppy to generate Fastq files. Alignment to the reference sequence, which contains 10 D4Z4 RUs and flanking sequences from 3950 bp upstream of CR1 to 251 bp downstream of CR4, was conducted using Minimap2. Reference sequences were constructed using SnapGene software (from Insightful Science; available at snapgene.com). For DNA methylation analysis, sense- and antisense-strand reads from the 4qA and 10q loci were re-aligned to the corresponding reference sequences and then Nanopolish was performed [18]. Reference sequences contained the detected size of D4Z4 RUs and flanking sequences from 327 bp downstream of CR2 to 1 bp upstream of CR3. Unipro UGENE free software and Integrative genomics viewer were used for sequence alignment [19, 20]. For analysis of correlation between the distal D4Z4 CpG methylation rate and clinical symptoms, we calculated mean CpG methylation rates of the most distal D4Z4 RUs (RU3, RU2, and the promoter region of RU1) for all 4qA-reads obtained from each FSHD1 sample. Mean methylation rate or D4Z4 length, and age at disease onset or age at hospital inspection were analyzed and plotted with Graphpad Prism, and correlation coefficients were calculated by linear regression.
Results
Determination of numbers of D4Z4 RUs in patients with facioscapulohumeral muscular dystrophy by Nanopore sequencing
For CAS9 cleavage, we designed two types of guide RNA each for the p13E-11 (CR1/CR2) and A-haplotype (CR3/CR4) regions; the distal guides, CR3 and CR4, specifically recognized the 4qA and 10q loci, but not 4qB (Fig. 1A, B). To validate the nCATS assay, we analyzed five samples (Sample 1–5) from patients genetically diagnosed with FSHD1 by Southern blotting (Tables 1 and 2). Reads derived from the 4qA locus were obtained after alignment to the reference sequence, and the number of D4Z4 RUs were calculated from the read length (Fig. 1C; red dots, Fig. 1D; Additional file 1: Table S1). Sample 1, 2, 3, 4, and 5 carried 1, 2, 3, 4, and 5 D4Z4 RUs, respectively, consistent with results from Southern blotting.

Determination of numbers of D4Z4 RUs in patients with facioscapulohumeral muscular dystrophy by Nanopore sequencing. A Schematic showing the D4Z4 repeat regions at the human chromosome 4qA, 4qB, and 10q loci. D4Z4 RUs are represented by triangles. The XapI and BlnI restriction enzyme sites are unique to chromosomes 4 and 10, respectively. The p13E-11, A-type haplotype, and B-type haplotype regions are indicated in green, blue, and pink, respectively. Green and blue arrows indicate crRNA cleavage sites (CR1/CR2/CR3/CR4). B The CR2 site in p13E-11 and CR3 site in the A-haplotype on the 4qA and 10q allele are shown by green and blue arrows, respectively. The 4qA polyadenylation signal is indicated in red. Sequences in the rectangle were used to distinguish reads from the 4qA and 10q loci. C Fragments carrying a single D4Z4 RU produced by CR2/CR3 or CR1/CR4 cleavage were 5.4 and 6.0 kb, respectively. D The length of identified reads and numbers of D4Z4 RUs are plotted. Red and black dots indicate reads derived from the 4qA and 10q loci, respectively
In addition to the 4qA locus, we also occasionally obtained reads from chromosome 10q in Samples 1 (13 RUs), 2 (13 RUs), and 3 (10 RUs and 12 RUs) (black dots in Fig. 1D and Additional file 1: Table S2). We confirmed that both the 4qA- and 10q-derived reads were correctly assigned by identifying 4qA-specific (XapI, Non-BlnI, and pA) and 10q-specific (Non-XapI, BlnI, and Non-pA) sequences, along with the common p13E-11 sequence (Additional file 1: Fig. S1). Moreover, we confirmed that identical results were obtained using genomic DNA samples from the same subject from different sources by comparing Samples 1 and 6. These results suggest that our method enables precise determination of the number of D4Z4 RUs and the haplotypes on which the repeats reside.
We also analyzed samples that were undiagnosed by Southern blotting following linear gel electrophoresis because we failed to detect 4qA-derived bands (Samples 7 and 8) or failed to determine repeat lengths based on restriction fragment sizes (Samples 9–15) (Table 2). Using nCATS, we successfully determined the repeat lengths of 4qA-derived reads even from these challenging samples, as follows: Sample 7, 11 RUs; Sample 8, 13 RUs; Sample 9, 4 RUs; Sample 10, 5 RUs; Sample 11, 5 RUs; Sample 12, 5 RUs; Sample 13, 3 RUs; Sample 14, 3 RUs; and Sample 15, 3 RUs (Fig. 1D).
A genomic deletion detected in patients with contracted D4Z4 repeats
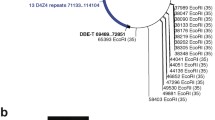
Interestingly, we also detected a genomic deletion, as an atypical cause of rearrangement of D4Z4 repeats. Samples 13 and 14 each generated one read with an intermediate size between 2 and 3 RUs. Sequence analysis revealed that both reads contained a deletion spanning 1.3 kb from 469 bases proximal to the most proximal D4Z4 RU to 859 bases within it (Fig. 2). Deletion within D4Z4 repeats has not been reported previously in FSHD1.

A genomic deletion detected in patients with contracted D4Z4 repeats. A Representative data showing reads obtained from Sample 13 mapped using Integrative Genomics Viewer. Among five reads (No. 1–5) with three repeat units (RUs) (11 kb), four reads (No. 1–4) with no deletion and one read (No. 5) with a 1.3 kb deletion were obtained. B The deletion was localized downstream of p13E-11 and extended to the middle of the most proximal D4Z4 RU
CpG methylation rates in D4Z4 RUs
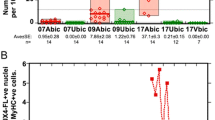
We also used nCATS results to determine the CpG methylation status of individual reads; therefore, we calculated CpG methylation rates for each RU in 4qA and 10q-derived reads (Fig. 3 and Additional file 1: Table S3). In FSHD1, the methylation rates of contracted 4qA-reads were consistently low, although those of the most distal D4Z4 RU at position 1 were relatively higher in most reads (Fig. 3). By contrast, the methylation rates of 10q-derived reads were low in proximal RUs, but elevated toward distal RUs. Further, in FSHD2, the CpG methylation rates of both 4qA- and 10q-reads were low throughout, with the exception of a few reads, in which the most distal RU1 was relatively highly methylated.

CpG methylation rates in D4Z4 RUs. CpG methylation rates of D4Z4 RUs in individual reads from the 4qA and 10q loci are plotted in red and black, respectively. D4Z4 RUs are numbered from the distal D4Z4 region
Methylation rates in the promoter region and gene body of the most distal D4Z4 RU
Next, we analyzed the CpG methylation rates of the promoter region and gene body of the most distal D4Z4 RU (RU1) separately (Fig. 4). Although Samples 1 and 6, which contained only one RU, showed similar CpG methylation rates in the promoter region and gene body, the methylation rates of promoter regions were generally lower than those in the gene body in all other samples from patients with both FSHD1 and FSHD2.

Methylation rates in the promoter region and gene body of the most distal D4Z4 RU. CpG methylation rates in the promoter and gene body of the most distal D4Z4 RU in individual reads are plotted. Reads from 4qA and 10q are shown in red and black, respectively
Correlation between CpG methylation rate in distal D4Z4 and patient phenotypes
Epigenetic changes in the contracted D4Z4 repeats on chromosome 4qA have been observed previously and are considered to be associated with the development of FSHD1 [10, 13, 14]. We hypothesized that the CpG methylation rate of the most distal D4Z4 RUs is a determinant of disease development; therefore, we examined the correlation between average methylation rate of the most distal three RUs (Fig. 3) and patient age at onset or at hospital inspection. As shown in Fig. 5, we found a moderate correlation between CpG methylation and age at onset (R2 = 0.645) than that between D4Z4 repeat length and age at onset (R2 = 0.401). Although the correlation coefficient between CpG methylation and age at hospital inspection was not high (R2 = 0.306), there was a tendency toward correlation, in that CpG methylation rate < 10% was associated with hospital inspection at a younger age (≤ 20 years old), while CpG methylation rates of 10–20% were associated with that at > 40 years old.

Correlation between CpG methylation rate in distal D4Z4 or repeat length, and patient phenotypes. A, C Scatter plots of mean CpG methylation rate in distal D4Z4 repeat units and age at disease onset (A) or age at hospital inspection (C) in FSHD1. B, D Scatter plots of D4Z4 repeat number and age at disease onset (B) or age at hospital inspection (D) in FSHD1. Correlation coefficients were calculated by linear regression. Two samples were removed due to unknown exact ages at onset of the disease
Discussion
In general, nCATS could be applicable to any other genetic disorders. In particular, it has an advantage on diagnosis of repeat-associated disorders, such as Huntington disease, spinal cerebellar ataxia, neuronal intranuclear inclusion disease, oculopharyngeal distal myopathy and others, in which the causative genetic variation cannot be identified by short read sequencing. In fact, it has been applied for analysis of some tandem repeat disorders, fragile X syndrome and myotonic dystrophy [21,22,23]. Nanopore sequencing was previously applied for analysis of FSHD using a bacterial artificial chromosome clone containing 13 D4Z4 repeat units [24].
In this study, we developed a direct sequencing system using nCATS to analyze clinical samples from patients with FSHD. Our method is more efficient and can collect more detailed information than conventional method. Conventional method for diagnosis of FSHD is carried out by multiple Southern blots for detection of the size of 4q-derived D4Z4 repeat and haplotyping 4q, and by bisulfite sequencing for measurement of the CpG methylation rate. In contrast, our method enables us to simultaneously identify the number and the methylation rate of D4Z4 repeat unit and the haplotype derived from 4qA. Our system has several advantages. First, long read sequencing can be applied to analysis of a similar DNA fragment size range to that detected by Southern blotting. Second, CRISPR/CAS9 enrichment allows barcoding sequencing of five samples simultaneously, saving time and cost. Third, single-molecule sequencing technology provides genetic information at the base level and can determine the number of RUs, even in samples that have mutated restriction enzyme sites, which prevent determination of RU number by the standard Southern blotting method. Finally, the nCATS system allows simultaneous detection of CpG methylation and D4Z4 RUs numbers, providing information about local epigenetic modification of D4Z4 repeats, due to the application of single-molecule sequencing of unamplified genomic DNA molecules derived from individual nuclei, without any bias.
Along with successful determination of D4Z4 RU numbers in patients, we also detected atypical rearrangement of D4Z4 repeats. As shown in Figs. 1D and 2, two reads of intermediate size had a 1.3 kb deletion in the most proximal D4Z4 RU, while p13E-11 was not deleted. This deletion is unlikely to be associated with the contraction of D4Z4 repeats in FSHD1, as the pathogenic alleles in FSHD1 usually maintain the intact RU structure, even when they contracted. Common atypical rearrangements found in individuals with FSHD1 have been reported, including D4Z4 proximally extended deletion (DPED1–7) alleles, which span 5.9–45.7 kb proximal to and within D4Z4, including p13E-11. In some DPED alleles, genetic elements, such as DUX4C, FRG2, DBE-T, and myogenic enhancers, are deleted, suggesting that their role in FSHD pathogenesis requires reevaluation [25].
The most important finding in our study was detection of DNA methylation rates across entire contracted and normal expanded D4Z4 repeat sequences from the 4qA and 10q loci. As shown in Fig. 3, 4qA-derived contracted reads were uniformly hypomethylated in patients with FSHD1, while both 4qA- and 10q-derived reads were uniformly hypomethylated in FSHD2, with the exception of a few reads. These results are similar to those generated in previous studies by Southern blot and bisulfite sequencing analyses [10, 13, 14], but our approach allows assessment of focal methylation rate at the nucleotide level. We further analyzed 10q-derived reads in FSHD1, and found that the methylation level was lower at proximal D4Z4 RUs (position 8–13), while it gradually increased (up to ≥ 60%) at distal RUs (positions 1–7). Given the mimicry of normal expanded 4qA-D4Z4 repeats by 10q-derived reads, these results suggest that only DNA hypermethylation at distal D4Z4 RUs contributes to suppression of the DUX4 gene in the normal 4qA allele, while contraction of D4Z4 repeats causes hypomethylation of distal D4Z4 similar to proximal D4Z4 in the 10q locus, leading to DUX4 expression and consequent development of FSHD1. Indeed, mean CpG methylation rate of the most distal RUs and disease onset in patients was well-correlated. A larger study of the relationships among methylation rate, D4Z4 contraction, and clinical phenotypes is needed. To this end, we aim to overcome the limitation of decreased acquisition of sequencing reads from alleles with more than 10 RUs.
Limitations
The nCATS method has limitations. First, the number of sequencing reads containing mildly contracted D4Z4 repeats (11–13 RUs) detected was quite low, particularly as only a few reads were obtained from the normal 10q locus, and no reads were obtained from some samples. The reasons why we could not obtain read from chromosome 10 in all samples and the number of reads in various samples are different are; (1) the difficulty to purify intact high molecular weight DNA, because the longer DNA might tend to be subject to degradation, (2) the difficulty to obtain longer DNA fragments beyond 13 RUs, because we used only the reads harboring full-length D4Z4 repeat in our analysis, (3) the efficacy of CAS9 cleavage of hypermethylated DNA, because distal D4Z4 were extremely higher methylation rates. Technical improvements in terms of preparation of genomic DNAs are required to overcome this shortcoming. Second, our method does not isolate reads derived from 4qB. Although the lack of analysis on 4qB is not likely to affect our conclusion, the epigenetic status in 4qB could be meaningful information as reference data for methylation rate of 4qA-derived D4Z4.
Conclusions
In this study, we successfully determined the hypomethylation of D4Z4 RUs in individual 4qA fragments in FSHD. The hypomethylation in the contracted D4Z4 in FSHD1 provides a good explanation why the shortening of D4Z4 repeats is associated with severe phenotypes in patients and it induces abnormal DUX4 expression which leads to developing FSHD. For a further improvement, we need to have a large cohort of patients and controls in the future, which might give a clue for complete understanding of the pathomechanism of FSHD.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional files.
Abbreviations
- FSHD:
-
Facioscapulohumeral muscular dystrophy
- RUs:
-
Repeat units
- nCATS:
-
Nanopore CRISPR/Cas9-targeted resequencing
References
Greco A, Goossens R, van Engelen B, van der Maarel SM. Consequences of epigenetic derepression in facioscapulohumeral muscular dystrophy. Clin Genet. 2020;97:799–814.
Lemmers RJLF, de Kievit P, Sandkuijl L, Padberg GW, van Ommen GJB, Frants RR, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6.
Lemmers RJLF, Van Der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3.
Snider L, Geng LN, Lemmers RJLF, Kyba M, Ware CB, Nelson AM, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6: e1001181.
Wijmenga C, Hewitt JE, Sandkuijp LA, Clark LN, Tracy J, Dauwerse WHG, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30.
Lemmers RJLF, Tawil R, Petek LM, Balog J, Block GJ, Santen GWE, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4.
Van Den Boogaard ML, Lemmers RJLF, Balog J, Wohlgemuth M, Auranen M, Mitsuhashi S, et al. Mutations in DNMT3B modify epigenetic repression of the D4Z4 repeat and the penetrance of facioscapulohumeral dystrophy. Am J Hum Genet. 2016;98:1020–9.
Hamanaka K, Šikrová D, Mitsuhashi S, Masuda H, Sekiguchi Y, Sugiyama A, et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology. 2020;94:e2441–7.
Surguchov A, Bernal L, Surguchev AA. Phytochemicals as regulators of genes involved in synucleinopathies. Biomolecules. 2021;11:624.
Van Overveld PGM, Lemmers RJFL, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7.
Haynes P, Bomsztyk K, Miller DG. Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenet Chromatin. 2018;11:47.
Zeng W, De Greef JC, Chen YY, Chien R, Kong X, Gregson HC, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1γ/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009;5: e1000559.
de Greef JC, Lemmers RJLF, van Engelen BGM, Sacconi S, Venance SL, Frants RR, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–59.
Jones TI, Yan C, Sapp PC, McKenna-Yasek D, Kang PB, Quinn C, et al. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin Epigenet. 2014;6:23.
Zampatti S, Colantoni L, Strafella C, Galota RM, Caputo V, Campoli G, et al. Facioscapulohumeral muscular dystrophy (FSHD) molecular diagnosis: from traditional technology to the NGS era. Neurogenetics. 2019;20:57–64.
Hartweck LM, Anderson LJ, Lemmers RJ, Dandapat A, Toso EA, Dalton JC, et al. A focal domain of extreme demethylation within D4Z4 in FSHD2. Neurology. 2013;80:392–9.
Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–4.
Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. 2017;14:407–10.
Okonechnikov K, Golosova O, Fursov M, Varlamov A, Vaskin Y, Efremov I, et al. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166–7.
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6.
Rasmussen A, Hildonen M, Vissing J, Duno M, Tümer Z, Birkedal U. High resolution analysis of DMPK hypermethylation and repeat interruptions in myotonic dystrophy type 1. Genes. 2022;13:970.
Alfano M, De Antoni L, Centofanti F, Visconti VV, Maestri S, DegliEsposti C, et al. Characterization of full-length CNBP expanded alleles in myotonic dystrophy type 2 patients by Cas9-mediated enrichment and nanopore sequencing. Elife. 2022;11: e80229.
Grosso V, Marcolungo L, Maestri S, Alfano M, Lavezzari D, Iadarola B, et al. Characterization of FMR1 repeat expansion and intragenic variants by indirect sequence capture. Front Genet. 2021;12: 743230.
Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H. Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep. 2017;7:14789.
Lemmers RJLF, Van Der Vliet PJ, Granado DSL, Van Der Stoep N, Buermans H, Van Schendel R, et al. High-resolution breakpoint junction mapping of proximally extended D4Z4 deletions in FSHD1 reveals evidence for a founder effect. Hum Mol Genet. 2022;31:748–60.
Acknowledgements
The authors would like to thank the patients, their families, and physicians who participated in the study.
Funding
This work was partly supported by an Intramural Research Grant for Neurological and Psychiatric Disorders of NCNP [3-9 to SN, 2-6 to SN, 2-5 to IN, SH]; AMED [22ek0109490h0003 to SN, SH, IN]; Nippon Shinyaku Research Grant [2021-Category B to YH], KAKENHI [21K15689 to YH], and the FSHD Society [FSHD-Fall2020-5973845533 to YH].
Author information
Authors and Affiliations
Contributions
Conceptualization: SN; formal analysis: YH, KI, YS, and SN; investigation: YH, YK, YS, and MO; methodology: YH and SN; patient evaluations, collecting patient samples, and/or clinical data: YS, MM, YO, YT, DK, NA, CM, TM, TH, KN, and KI; visualization: YH, YS, and SN; software: KI; resources: YG and IN; supervision: SH, SN, and IN; project administration: SN; funding acquisition: YH, SH, SN, and IN; writing—original draft: YH and SN; writing—reviewed: all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Samples and data were collected between January 1978 and December 2021 from the National Center of Neurology and Psychiatry registry. Fourteen patients were selected, of whom five had 1, 2, 3, 4, or 5 D4Z4 RUs, while data were inconsistent for seven patients, and two patients showed no bands on linear Southern blotting of genomic DNA samples extracted from peripheral blood lymphocytes. The oldest clinical description available for each patient (data at hospital inspection) was reviewed. Clinical characteristics and the results of Southern blotting are described in Tables 1 and 2, respectively. Fibroblasts from Patient 1 were obtained from the NCNP Biobank. This study was approved by the ethics committee of the National Center of Neurology and Psychiatry, Japan.
Consent for publication
Materials used in this study were obtained for diagnostic purposes with written informed consent. All participants were enrolled after providing informed consent.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Characteristic sequences detected by nCATS. Sequences of representative (A) 4qA- and (B) 10q-derived reads obtained from the indicated samples. The XapI/non-XapI and BlnI/non-BlnI sites in the most distal D4Z4 RU are shown. In Samples 8, 14, and 15, the XapI, XapI and non-BlnI, and non-XapI sites, respectively, in the second most distal D4Z4 RU are shown, due to the difficulty in identifying restriction sites. Table S1. Lengths of reads derived from the 4qA locus in each patient. Table S2. Lengths of reads derived from the 10q locus in each patient. Table S3. Methylation rates across all D4Z4 RUs at the 4qA and 10q loci.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hiramuki, Y., Kure, Y., Saito, Y. et al. Simultaneous measurement of the size and methylation of chromosome 4qA-D4Z4 repeats in facioscapulohumeral muscular dystrophy by long-read sequencing. J Transl Med 20, 517 (2022). https://doi.org/10.1186/s12967-022-03743-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-022-03743-7