
Abstract
Background
Noninvasive prenatal screening (NIPS) based on cell-free fetal DNA (cffDNA) has rapidly been applied into clinic. However, the reliability of this method largely depends on the concentration of cffDNA in the maternal plasma. The chance of test failure results or false negative results would increase when cffDNA fraction is low. In this study, we set out to develop a method to enrich the cffDNA for NIPS based on the size difference between cell-free DNA (cfDNA) of fetal origin and maternal origin, and to evaluate whether the new NIPS method can improve the test quality.
Methods
We utilized 10,000 previous NIPS data to optimize a size-selection strategy for enrichment. Then, we retrospectively performed our new NIPS method with cffDNA enrichment on the 1415 NIPS samples, including 1404 routine cases and 11 false negative cases, and compared the results to the original NIPS results.
Results
The 10,000 NIPS data revealed the fetal fraction in short cfDNA fragments (< 160 bp) is significantly higher. By using our new NIPS strategy on the 1404 routine cases, the fetal fraction increased from 11.3 ± 4.2 to 22.6 ± 6.6%, and the new method performed a significant decrease of test-failure rate (0.1% vs 0.7%, P < 0.01). Moreover, in 45.5% (5/11) of the false negative cases, fetal trisomies were successfully detected by our new NIPS method.
Conclusions
We developed an effective method to enrich cffDNA for NIPS, which shows an increased success rate and a reduced chance of false negative comparing to the ordinary NIPS method.
Similar content being viewed by others


Background
The discovery of cell-free fetal DNA (cffDNA) in maternal plasma by Lo et al. in 1997 has inspired various non-invasive prenatal screening (NIPS) applications [1], which avoids the ~ 1:100 chance for miscarriage introduced by invasive sampling. At present, NIPS for common fetal aneuploidies, based on analysis of cffDNA in maternal plasma, has been gradually applied as a first-tier aneuploidy screening strategy in clinical practice [2, 3]. Previous large-scale clinical studies have revealed high accuracy of NIPS in screening on trisomy 21, 18 and 13, with sensitivity and specificity higher than 95% [4,5,6].
Importantly, the reliability of NIPS largely depends on the assumption that there is sufficient fetal DNA in the samples tested [7]. Fetal fraction, the percentage of cell-free DNA (cfDNA) that is from fetal origin in maternal peripheral blood, is generally at the range of 3–30%, with an average of about 13% [4]. The cffDNA levels are determined by multiple factors, including gestational age, maternal weight and extraction method [8, 9]. In addition, fetal fraction could further decrease during sample transportation or laboratory work-up in NIPS. Previous researches have shown that the extent of chromosomal abnormalities presented in plasma of women with aneuploidy pregnancies is linearly correlated with the cffDNA fraction, thus the test accuracy of NIPS largely relies on the fetal fraction [10]. Most current NIPS protocols utilize 4% as the lower fetal fraction cutoff value to ensure a reliable result. However, for pregnancies at early GA stages or obese women who require NIPS, low fetal fraction is the major issue to overcome [8]. In addition, NIPS misses about 1% chromosomal aneuploidy cases, and the most common factor associated with these false negative results is the low fetal fraction [11, 12]. As for the aforementioned reasons, it is critical to elevate fetal fraction for achieving convincing NIPS results.
cfDNA is DNA fragments generated from apoptotic cells, which is released into circulation after rapid DNA degradation. The size distribution of these DNA fragments has peaks corresponding to nucleosomes (~ 143 bp) and chromatosomes (nucleosome + linker histone; ~ 166 bp) [13, 14]. In pregnant women, cffDNA in maternal peripheral blood mainly originates from placental trophoblasts, crossing through the placental barrier [1]. In 2010, Lo et al. found that cffDNA exhibits a different length distribution comparing to the cfDNA from maternal cells, with a reduced proportion of molecules of 166 bp and an increased proportion of molecules of shorter than 150 bp in maternal plasma, possibly caused by differential nucleosomal packaging during apoptosis, or by differences in the force of nucleosome binding [15]. Based on these findings, it is theoretically possible to enrich cffDNA fragments from total cfDNA in maternal peripheral blood by size selection.
In the present study, we set out to develop an experimental method for cffDNA enrichment, which increased the mean cffDNA fraction by 1.5–4 times, while the complexity of cfDNA obtained is fully adequate for NIPS. Moreover, we retrospectively tested 1415 clinical samples including 1404 routine clinical NIPS samples and 11 false negative NIPS samples using this new method. Our results demonstrated the cffDNA enrichment strategy can improve the overall performance of NIPS by reducing false negative results as well as the test failure rate.
Methods
DNA extraction and libraries construction
Five to 10 mL of peripheral venous blood was collected from each participating pregnant woman in EDTA-containing tubes or Streck blood collection tubes. The blood samples were first centrifuged at 1600×g for 10 min at 4 °C to separate the plasma from peripheral blood cells. The plasma portion was carefully transferred to a polypropylene tube and subjected to centrifugation at 16,000×g for 10 min at 4 °C to pellet the remaining cells. Cell-free DNA from 600 μL of maternal plasma was extracted using the QIAamp DSP DNA Blood Mini Kit (Qiagen) following the blood and body fluid protocol. End-repairing, adaptor ligation and PCR amplification were performed using Ion Plus Fragment Library Kit (Life Technologies).
Cell-free fetal DNA enrichment
DNA enrichment was performed after end-repairing and before adaptor ligation during NIPS library construction. Magnetic beads with an average particle size of 1 μm were used for the purpose of size-selecting the end-repaired DNA fragments with size smaller than 160 bp. To achieve the highest efficiency, we optimized this step by testing a series of different bead concentrations. Magnetic beads were added to the end-repaired DNA fragments, followed by vibrating the tubes for at least 3 s. The tubes were then suspended for 5 min and transferred to a magnetic rack. The supernatant containing size-selected DNA fragments was then transferred to another tube for adaptor ligation.
Sequencing
DNA library concentration was determined by Qubit and RT-PCR. For DNA sequencing, 15–20 libraries were pooled and sequenced using JingXin BioelectronSeq 4000 System (CFDA registration permit NO. 20153400309) semiconductor sequencer with single-end sequencing mode in 400 flows producing raw sequencing reads with size of up to 200 bp and counts of at least 3.5 million.
Data analysis
Reads trimmed from the 3′ end by sequencing quality value of > 15 and filtering by reads with length of shorter than 50 bp were aligned to the human genomic reference sequences (hg19) using the BWA [16]. Reads that were unmapped or had multiple primary alignment records were filtered by FLAG field in the alignment file, using an in-house Perl script. Duplicate reads were identified by Picard (http://picard.sourceforge.net/). The remaining reads were considered unique reads for further analysis. To eliminate the effect of GC bias, we calculated the number of unique reads for each 20 kb-bin, then applying an integrated method for GC correction using a three-step process: Locally weighted scatterplot smoothing (LOESS) regression [17], intrarun normalization [18], and linear model regression [19]. LOESS regression was performed in R software with default parameters. We derived a z score for each of the chromosomes in a test sample by subtracting the mean chromosome ratio in a reference set of euploid control pregnancies from the chromosome ratio in a test case and dividing by the SD of the chromosome ratio in the reference set according to the following equation: a cutoff value of z score > 3 was used to determine whether the ratio of the chromosome was increased and hence fetal trisomy was present.
Estimation of cffDNA fraction
Two types of methods were used to calculate the fetal DNA fraction in maternal plasma. For one method, the cffDNA fraction for pregnancy with a male fetus can be easily estimated using reads proportion on the Y chromosome. For the other method, the cffDNA fraction can be estimated using length distribution of cffDNA. Fetal DNA is generally shorter than maternal DNA, Plasma samples with a higher fetal DNA fraction would have a higher proportion of short plasma DNA fragments (~ 130–140 bp; region A) and a lower proportion of long plasma DNA fragments (~ 155–175 bp; region B). LOESS regression was applied to fit the fetal fraction against reads ratio in features A and B. We obtained the LOESS fit-predicted fetal fraction PA for feature A and PB for feature B. Because both PA and PB predict the fetal DNA fraction, PA and PB should also closely correlate. Therefore, we used reference samples to compare PA and PB and thus, identify instances of poor correlation. If Pdiff = (PA − PB) × 2/(PA + PB) is larger than 0.40 (larger than 99% normal samples), PA and PB are inconsistent, and the fetal fraction is considered unpredictable. Otherwise, we calculated the final predicted fetal fraction using P = (PA + PB)/2.
DNA complexity calculation
In order to evaluate the DNA loss during the enrichment procedure, the amounts of sequencing libraries with or without enrichment were calculated using qPCR and KAPA Library Quantification Kit. The qPCR with primers of 5′-CCTCTCTATGGGCAGTCGGTG-3′ and 5′-CCTGCGTGTCTCCGACTCAG-3′ was performed using SYBR Green Realtime PCR Master Mix (TOYOBO). Besides the amounts, DNA complexity was a major factor for PCR amplification and sequencing. Lower DNA complexity would result with higher percentage of duplicated reads when sequencing, leading to the test failures of NIPS. Thus, we applied a method of captured sequencing covering 300 SNPs for libraries constructed with and without beads enrichment. Capture probes (data not provided) were designed and synthetized by Agilent. DNA hybridization and sequencing were performed according to the manufacturer’s instructions. After reads mapping and SNP calling, DNA complexities were calculated using the following equation:
NIPS sample cohort
10,000 NIPS data, 1404 regular NIPS cases and 11 false negative cases were recruited in this study. All participants signed informed written consent before blood collection. This study was approved by the institutional review board of the Affiliated Obstetrics and Gynecology Hospital of Nanjing Medical University.
Confirmation of original NIPS results
Pregnancies with positive NIPS results were recommended for confirmatory invasive prenatal diagnosis using amniocentesis following karyotyping and/or chromosomal microarray analysis (CMA). Pregnancies with negative NIPS results were interviewed at 3 months after delivery to record the information, including the ultrasound examination, pregnancy outcomes, newborn physical examination results, and neonatal/fetal cytogenetic analysis.
Statistical analysis
Statistical analysis between the different groups was performed using a Chi square (X2) test or Fisher’s exact test, and P-values of ≤ 0.01 were considered statistically significant.
Results
Optimizing size-selection strategy for cffDNA enrichment
Based on the hypothesis that the fetal fraction are correlated with size distribution of the cfDNA fragments, we set out to develop a method that can improve cffDNA fraction by selectively enriching short cfDNA fragments.
In order to investigate the correlation between cfDNA fragment size and cffDNA fraction, we collected a total of 10,000 existing NIPS data from pregnancies with male fetus. We grouped data from every 100 samples into one mixed data set, generating a total of 100 mixed data sets. The data set was produced by single-end sequencing mode with read length of 200 bp and read counts of 3.5 million using JingXin BioelectronSeq 4000 System. For each mixed data set, reads were divided into 10 different bins according to their sequence length, including [100, 110), [110, 120), [120, 130), [130, 140), [140, 150), [150, 160), [160, 170), [170, 180), [180, 190) and [190, 200) (Fig. 1a). The portion of chromosome Y reads, representing the fetal fraction, were calculated for each bin, respectively, which were then compared to that in the pooled data of the 10,000 NIPS results. In bins of [100, 110), [110, 120), [120, 130), [130, 140), [140, 150) and [150, 160), the mean fetal fractions were shown to increase by 1.98, 2.42, 2.64, 2.59, 2.01 and 1.44 times comparing to the average fetal fraction in pooled data. These results demonstrate in cfDNA fragments < 160 bp, the fetal fraction is significantly higher than that in the total cfDNA fragments in the maternal peripheral blood. As a result, it is possible to increase the cffDNA fraction by analyzing only sequences < 160 bp.

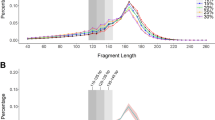
The effect of cffDNA enrichment. a Foldchange of cffDNA fraction in 10 different bins, including [100, 110), [110, 120), [120, 130), [130, 140), [140, 150), [150, 160), [160, 170), [170, 180), [180, 190), [190, 200), calculated using reads portion of the Y chromosome. b Alteration of read length distribution after size-selection using our customized beads with different dosages. c The portion of short reads (< 160) increased after size-selection using our customized beads with different dosages. d The Z-score of ChrY increased after size-selection using our customized beads with different dosages
In order to achieve cffDNA enrichment, we developed our customized beads with three different concentrations (1.2×, 1.3× and 1.5× ratio) to samples. To evaluate the effect of our enrichment procedure, 174 blood samples from pregnancies with male fetus were tested using our enrichment procedure, and the portions of short reads (< 160 bp), long reads (≥ 160 bp) and the chromosome Y reads were calculated. Among them, 55 samples, 74 samples and 45 samples were processed using 1.2×, 1.3× and 1.5× ratio of beads, respectively. While the other 133 samples were processed without beads enrichment. Sequencing results revealed the distribution of read lengths altered (Fig. 1b). The portion of short DNA fragment (< 160 bp) was significantly increased using the beads with all three concentration (Fig. 1c). Importantly, the reads ratio of chromosome Y were also increased by 1.5–4 times, most significantly in beads with 1.5× ratio (Fig. 1d). These results proved that our enrichment procedure using 1.5× ratio of beads can effectively increase the portion of cffDNA comparing to that without enrichment
Since the procedure of cffDNA enrichment would discard most long cfDNA fragments, we quantified the amounts of sequencing libraries before and after enrichment, and found an approximately 91.76% reduction of input cfDNA would occur, which is a major concern of this method. For this reason, we tested whether the reduction of input DNA could impair the original library complexity, which represented the copies of whole genome DNA in cfDNA. Duplicated reads were considered as a good evaluation index to assess the robustness of the library. Lower rate of duplication represented higher complexity of input DNA, leading to a more reliable NIPS result. In order to find out the minimal threshold of input DNA complexity, we performed a computer simulation: first, we built a virtual input DNA pool with 0.1 to 100 copies of whole genome DNAs (library complexity ranged from 0.1 to 100). Then, we randomly sampled the reads with the size of 150 bp, which is close to the average cfDNA fragment size in maternal plasm, from virtual input DNA pools in different sequencing depth, ranged from 0.1× to 1×. Finally, we counted the duplication rate in different combinations of input DNA complexity and sequencing depth. The result revealed a low duplication rate with limited variation can be achieved with sequencing depth of 0.1× when input DNA complexity is higher than 3, which can be considered as the minimal input DNA complexity threshold for NIPS (Additional file 1: Figure S1). Furthermore, we developed a SNP-based method to compute the DNA complexity on 12 plasma samples (600 μL) before and after cffDNA enrichment. Although the DNA complexity reduced from 323.69 ± 77.42 to 84.09 ± 61.37 after enrichment, this number is still significantly higher than the threshold of required complexity of 3 in all the samples tested (Additional file 2: Table S1). In all, we proved that although cffDNA enrichment could reduce the input DNA complexity, our procedure can still obtain fully adequate DNA from maternal plasma for performing NIPS.
Performance of NIPS using cffDNA enrichment on 1404 clinical samples
To evaluate the performance of NIPS with cffDNA enrichment, we recruited and restored plasma samples from 1404 women with singleton pregnancies who chose to have NIPS from Jan. 2017 to Feb. 2017 in our center. The clinical information is summarized in Additional file 3: Table S2. We retrospectively performed NIPS with cffDNA enrichment on these samples. We also collected the original ordinary NIPS results, as well as the confirmatory diagnostic results and the follow-up information, for further comparison.
We first selected the 902 samples with male fetuses from the 1404 samples and calculated their fetal fractions by reads ratio of chromosome Y. Original NIPS results revealed an average cffDNA fraction of 11.3 ± 4.2%, while the fetal fraction from the results of NIPS with cffDNA enrichment increased to 22.6 ± 6.6% (Fig. 2). To testify whether the elevated fetal fraction ensures a better performance on clinical samples, we compared the 1404 results of the new NIPS with cffDNA enrichment to the original NIPS results, as well as the confirmatory diagnostic results and the follow-up information. These results were summarized in Table 1. In brief, the original NIPS results showed a sensitivity of 100% (5/5), a specificity of 99.8% (1394/1397) and a positive predictive value (PPV) of 62.5% (5/8). While the new NIPS method showed a sensitivity of 100% (5/5), a specificity of 99.6% (1394/1399) and a PPV of 50% (5/10) (Table 1). Notably, the test-failure rate was significantly reduced in the new NIPS results (0.1% vs 0.7%, P < 0.01) (Additional file 4: Table S3).

Comparison between the cffDNA fractions from new NIPS method and the ordinary method tested on the 902 pregnancies with male fetus. a cffDNA fractions of the 902 samples using the two NIPS methods. Blue dots represented the sorted cffDNA fractions from ordinary NIPS, while red dots represent that of the corresponding samples using NIPS method with enrichment. b Boxplot for cffDNA fraction between the two NIPS methods
Performance of NIPS using cffDNA enrichment on 11 false negative samples
In clinic, the cause of false negative in NIPS is closely associated with low fetal fraction. To testify whether the new method could avoid the false negative results, a total of 11 restored false negative plasma samples from more than routine 100,000 ordinary NIPS cases were collected. By using NIPS with cffDNA enrichment, the fetal fractions were significantly increased by 1.9–2.7 times (P < 0.01). Importantly, 5 out of the 11 samples (45%) returned a positive result using the new method (Table 2), and these 5 positive results were consistent with the confirmatory karyotyping results of the fetuses. Our data demonstrated the cffDNA enrichment can effectively reduce the false negative rate of NIPS.
Discussion
In this study, we developed a working experimental method for cffDNA enrichment, which can effectively increase the mean cffDNA fraction and obtain fully adequate cfDNA for NIPS. By retrospectively performing our new method on the 1415 clinical samples including 1404 routine clinical NIPS samples and 11 false negative NIPS samples, we were able to improve the test quality in the way of reducing test-failure rate and false negative rate.
To date, several attempts have been reported for fetal cfDNA enrichment in NIPS technology. Qiwei Yang et al. reported a PCR-based enrichment method to selectively amplify the fetal cfDNA [20]. Another work from Joaquim Vong et al. reported a single-strand DNA library preparation method to enrich the short cfDNA in maternal plasma [21]. Moreover, Stephanie Yu et al. reported a size-based method instead of count-based method for NIPS recently, which also showed a promising ability in detecting common trisomies [22]. These works all focused on the size difference of the cffDNA in maternal plasma to make progress in NIPS. However, evaluation on the feasibility of these methods still need validation by sufficient clinical samples. In this study, to ensure a reliable result, clinical samples with detailed follow-up information, including the fetal karyotype/CMA results or postnatal interview, were used to validate our new NIPS method.
The existence of discordant results, including false positive and false negative, has been regarded as one of the major limitations in NIPS [23]. In clinic, invasive confirmatory diagnosis will be recommended to all the NIPS positive cases to minimize the adverse effects of false positive results. While the false negative cases would often cause harmful consequences. Therefore, it is critical to uncover the underlying mechanisms of these false negatives results and improve the testing method accordingly. It is widely accepted that the false negative NIPS result is closely associated with low fetal fraction and true fetal mosaicism (TFM) [23]. Although most current NIPS protocol set up a fetal fraction threshold of 4% for the reliable testing result, there is still a chance of false negative when the fetal fraction is low, such as slightly higher than 4%. As suggested in the previous literature, all false negative NIPS cases are recommended to undergo fetal cfDNA enrichment, which can help to identify low fetal fraction as the potential cause [24]. Our results showed about half of the false negative cases (5/11) could be avoided by the new NIPS method. Interestingly, the 5 corrected false negative samples have relatively lower fetal fractions (4.1–6.4%) than the other 6 samples (7.1–12.4%), which also indicated the false negative results in these 5 cases could be due to the low fetal fraction. While the other 6 false negative cases may associate with other causes, such as TFM. However, the placental tissue, which is not available in these cases, is required for the confirmation of TFM.
By using our method with cffDNA enrichment, we were able to avoid 9 test-failure results, compared to ordinary NIPS method on the 1404 clinical samples. Importantly, our clinical follow-up information confirmed those cases with original test-failure results were all true negative. In clinical circumstance, cases with ‘no-call’ result were recommended a blood re-draw and another test, but no successful result can be guaranteed. Avoiding test-failures will result in a shorter turnaround time as well as a lower cost for the test, which should also generate less anxiety for the women.
One downside of our strategy is the elevated overall false-positive rates. In this study, there were 2 additional false positive cases among the results of the 1404 clinical samples. As confined placental mosaicism is considered as the major factor causing false positive [23], the placental chromosomal abnormalities could be amplified with cffDNA enrichment. However, our results indicated the decreased specificity (99.8% vs 99.6%) and increased false positive rate (0.6% vs 0.7%) in the test cohort is limited, and still comparable to other studies [25]. A larger scale validation of this method would be favorable in the future. Another disadvantage is that the original fetal fraction information would be lost using the new method. It is reported the level of fetal fraction could be related to some pregnancy complications, such as spontaneous preterm delivery, intrauterine growth retardation (IUGR) and pre-eclampsia in asymptomatic pregnant women, suggesting its potential diagnostic value [26,27,28]. While after fetal cfDNA enrichment, the original fetal fraction information is no longer available. Although this is beyond the scope of this study, further research may be addressed to determine whether there is any association between the original fetal fraction, the enriched fetal fraction and pregnancy complications.
It is important to accurately estimate the fetal fraction in NIPS. Previously, cffDNA levels were examined by qPCR [1]. When massive parallel sequencing is performed on both the maternal and cffDNA in a given plasma sample, the fetal fraction can be determined by examining genetic elements that differ between maternal and fetal DNA [29], including Y chromosomal markers [30], polymorphic markers [31], DNA methylation markers [32], and size-, count- and nucleosome profile-based methods [22, 33]. Genes on the Y chromosome are the most commonly used distinguishing marker [11, 12], but this method can be only used in pregnancies with male fetuses. Of note, cfDNA in maternal plasma is readily digested into small fragments by natural processes. Because of the small size of these fragments, no additional shearing is required before sequencing. Meanwhile, the fetal and maternal derived DNA fragments exhibit a difference in the distributions of size peaks [15]. Based on these characters, size-based estimation of fetal DNA fraction was established for the pregnancies with both male and female fetuses, that was also performed in this and our previous paper [34].
Although it is still controversial to expand the scope of NIPS at present, the technology to use cfDNA for detecting fetal copy number variations (CNVs) and single gene disorders has been developed for years. Several groups utilized whole genome sequencing, SNP-based and targeted sequencing of maternal plasma DNA and showed its huge potential for the detection of fetal microdeletion/microduplication syndromes [35,36,37,38]. However, the core statistical procedure is to compare the reads dosage on the target region between testing sample and normal controls, which is highly dependent on the cffDNA fraction. Previous studies showed that the minimal cffDNA fraction requirement for this purpose was 10% [34]. In addition, the cfDNA-based NIPS for single-gene disorders is much more challenging, because the cfDNA in maternal plasma is generally of minor population, hampering the reliable deduction of the maternal inherence of pathogenic variants at single-nucleotide resolution. Technologically, the development of relative haplotype dosage analysis (RHDO), which utilizes information regarding parental haplotypes flanking the variants of interest, has been demonstrated to greatly improve the accuracy of single-gene disorder detection [39]. Therefore, our method of cffDNA enrichment could contribute in expanding NIPS to the prenatal detection of fetal CNVs and single gene disorders in the future.
Conclusions
This study demonstrates that it is feasible to increase the cffDNA fraction by selectively enriching short cfDNA fragments. Although the use of cffDNA enrichment in NIPS slightly decreased the specificity, this new method can avoid most test-failure results and reduce near half of false negative results caused by low fetal fraction, which improves the overall performance of NIPS in clinic. Our data also suggests the feasibility of using NIPS in detecting CNVs and single gene disorders in the future.
Abbreviations
- NIPS:
-
noninvasive prenatal screening
- cffDNA:
-
based on cell-free fetal DNA
- cfDNA:
-
cell-free DNA
- LOESS:
-
locally weighted scatterplot smoothing
- CMA:
-
chromosomal microarray analysis
- PPV:
-
positive predictive value
- TFM:
-
true fetal mosaic
- IUGR:
-
intrauterine growth retardation
- CNVs:
-
copy number variations
- RHDO:
-
relative haplotype dosage analysis
References
Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–7.
Song Y, Huang S, Zhou X, Jiang Y, Qi Q, Bian X, Zhang J, Yan Y, Cram DS, Liu J. Non-invasive prenatal testing for fetal aneuploidies in the first trimester of pregnancy. Ultrasound Obstet Gynecol. 2015;45:55–60.
Kagan KO, Sonek J, Wagner P, Hoopmann M. Principles of first trimester screening in the age of non-invasive prenatal diagnosis: screening for chromosomal abnormalities. Arch Gynecol Obstet. 2017;296:645–51.
Hu H, Liu H, Peng C, Deng T, Fu X, Chung C, Zhang E, Lu C, Zhang K, Liang Z, Yang Y. Clinical experience of non-invasive prenatal chromosomal aneuploidy testing in 190,277 patient samples. Curr Mol Med. 2016;16:759–66.
Mackie FL, Hemming K, Allen S, Morris RK, Kilby MD. The accuracy of cell-free fetal DNA-based non-invasive prenatal testing in singleton pregnancies: a systematic review and bivariate meta-analysis. BJOG. 2017;124:32–46.
Liang D, Lin Y, Qiao F, Li H, Wang Y, Zhang J, Liu A, Ji X, Ma D, Tao J, et al. Perinatal outcomes following cell-free DNA screening in > 32,000 women: clinical follow-up data from a single tertiary center. Prenat Diagn. 2018. https://doi.org/10.1002/pd.5328.
Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE. The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat Diagn. 2013;33:667–74.
Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell-free DNA at 11–13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013;41:26–32.
Jorgez CJ, Dang DD, Simpson JL, Lewis DE, Bischoff FZ. Quantity versus quality: optimal methods for cell-free DNA isolation from plasma of pregnant women. Genet Med. 2006;8:615–9.
Xu XP, Gan HY, Li FX, Tian Q, Zhang J, Liang RL, Li M, Yang XX, Wu YS. A method to quantify cell-free fetal DNA fraction in maternal plasma using next generation sequencing: its application in non-invasive prenatal chromosomal aneuploidy detection. PLoS ONE. 2016;11:e0146997.
Chiu RW, Chan KC, Gao Y, Lau VY, Zheng W, Leung TY, Foo CH, Xie B, Tsui NB, Lun FM, et al. Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma. Proc Natl Acad Sci USA. 2008;105:20458–63.
Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc Natl Acad Sci USA. 2008;105:16266–71.
Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57.
Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487:320–4.
Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, Zheng YW, Leung TY, Lau TK, Cantor CR, Chiu RW. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:61ra91.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–7.
Jiang F, Ren J, Chen F, Zhou Y, Xie J, Dan S, Su Y, Xie J, Yin B, Su W, et al. Noninvasive Fetal Trisomy (NIFTY) test: an advanced noninvasive prenatal diagnosis methodology for fetal autosomal and sex chromosomal aneuploidies. BMC Med Genomics. 2012;5:57.
Chen EZ, Chiu RW, Sun H, Akolekar R, Chan KC, Leung TY, Jiang P, Zheng YW, Lun FM, Chan LY, et al. Noninvasive prenatal diagnosis of fetal trisomy 18 and trisomy 13 by maternal plasma DNA sequencing. PLoS ONE. 2011;6:e21791.
Yang Q, Du Z, Song Y, Gao S, Yu S, Zhu H, Ren M, Zhang G. Size-selective separation and overall-amplification of cell-free fetal DNA fragments using PCR-based enrichment. Sci Rep. 2017;7:40936.
Vong JSL, Tsang JCH, Jiang P, Lee WS, Leung TY, Chan KCA, Chiu RWK, Lo YMD. Single-Stranded DNA library preparation preferentially enriches short maternal DNA in maternal plasma. Clin Chem. 2017;63:1031–7.
Yu SC, Chan KC, Zheng YW, Jiang P, Liao GJ, Sun H, Akolekar R, Leung TY, Go AT, van Vugt JM, et al. Size-based molecular diagnostics using plasma DNA for noninvasive prenatal testing. Proc Natl Acad Sci USA. 2014;111:8583–8.
Hartwig TS, Ambye L, Sorensen S, Jorgensen FS. Discordant non-invasive prenatal testing (NIPT)—a systematic review. Prenat Diagn. 2017;37:527–39.
Pescia G, Guex N, Iseli C, Brennan L, Osteras M, Xenarios I, Farinelli L, Conrad B. Cell-free DNA testing of an extended range of chromosomal anomalies: clinical experience with 6388 consecutive cases. Genet Med. 2017;19:169–75.
Zhang H, Gao Y, Jiang F, Fu M, Yuan Y, Guo Y, Zhu Z, Lin M, Liu Q, Tian Z, et al. Non-invasive prenatal testing for trisomies 21, 18 and 13: clinical experience from 146,958 pregnancies. Ultrasound Obstet Gynecol. 2015;45:530–8.
Quezada MS, Francisco C, Dumitrascu-Biris D, Nicolaides KH, Poon LC. Fetal fraction of cell-free DNA in maternal plasma in the prediction of spontaneous preterm delivery. Ultrasound Obstet Gynecol. 2015;45:101–5.
Al Nakib M, Desbriere R, Bonello N, Bretelle F, Boubli L, Gabert J, Levy-Mozziconacci A. Total and fetal cell-free DNA analysis in maternal blood as markers of placental insufficiency in intrauterine growth restriction. Fetal Diagn Ther. 2009;26:24–8.
Rolnik DL, O’Gorman N, Fiolna M, van den Boom D, Nicolaides KH, Poon LC. Maternal plasma cell-free DNA in the prediction of pre-eclampsia. Ultrasound Obstet Gynecol. 2015;45:106–11.
Peng XL, Jiang P. Bioinformatics approaches for fetal DNA fraction estimation in noninvasive prenatal testing. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18020453.
Lun FM, Chiu RW, Chan KC, Leung TY, Lau TK, Lo YM. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–72.
Nygren AO, Dean J, Jensen TJ, Kruse S, Kwong W, van den Boom D, Ehrich M. Quantification of fetal DNA by use of methylation-based DNA discrimination. Clin Chem. 2010;56:1627–35.
Jensen TJ, Kim SK, Zhu Z, Chin C, Gebhard C, Lu T, Deciu C, van den Boom D, Ehrich M. Whole genome bisulfite sequencing of cell-free DNA and its cellular contributors uncovers placenta hypomethylated domains. Genome Biol. 2015;16:78.
Kim SK, Hannum G, Geis J, Tynan J, Hogg G, Zhao C, Jensen TJ, Mazloom AR, Oeth P, Ehrich M, et al. Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenat Diagn. 2015;35:810–5.
Yin AH, Peng CF, Zhao X, Caughey BA, Yang JX, Liu J, Huang WW, Liu C, Luo DH, Liu HL, et al. Noninvasive detection of fetal subchromosomal abnormalities by semiconductor sequencing of maternal plasma DNA. Proc Natl Acad Sci USA. 2015;112:14670–5.
Srinivasan A, Bianchi DW, Huang H, Sehnert AJ, Rava RP. Noninvasive detection of fetal subchromosome abnormalities via deep sequencing of maternal plasma. Am J Hum Genet. 2013;92:167–76.
Lefkowitz RB, Tynan JA, Liu T, Wu Y, Mazloom AR, Almasri E, Hogg G, Angkachatchai V, Zhao C, Grosu DS, et al. Clinical validation of a noninvasive prenatal test for genomewide detection of fetal copy number variants. Am J Obstet Gynecol. 2016;215:227 e1–16.
Gross SJ, Stosic M, McDonald-McGinn DM, Bassett AS, Norvez A, Dhamankar R, Kobara K, Kirkizlar E, Zimmermann B, Wayham N, et al. Clinical experience with single-nucleotide polymorphism-based non-invasive prenatal screening for 22q11.2 deletion syndrome. Ultrasound Obstet Gynecol. 2016;47:177–83.
Neofytou MC, Tsangaras K, Kypri E, Loizides C, Ioannides M, Achilleos A, Mina P, Keravnou A, Sismani C, Koumbaris G, Patsalis PC. Targeted capture enrichment assay for non-invasive prenatal testing of large and small size sub-chromosomal deletions and duplications. PLoS ONE. 2017;12:e0171319.
Hudecova I, Chiu RW. Non-invasive prenatal diagnosis of thalassemias using maternal plasma cell free DNA. Best Pract Res Clin Obstet Gynaecol. 2017;39:63–73.
Authors’ contributions
PH, DL and ZX designed the experiments. PH, DL, YL, HLi and YC carried out the experiments. DL and HL wrote the manuscript. HL and ZX supervised the project. DL and YL analyzed the data, FQ, TW, CP and DLuo collected the samples. All authors provided critical comments and editorial modifications. All authors read and approved the final manuscript.
Acknowledgements
We would like to acknowledge Fengying Wang and Suqin Zheng for assistance in the collection of the follow-up information.
Competing interests
Y.C., C.P., D.Luo, and H.L. are employees of CapitalBio Technology Inc. The other authors declare no competing interest.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study was approved by the institutional review board of Obstetrics and Gynecology Hospital Affiliated to Nanjing Medical University.
Funding
This work was supported by the National Natural Science Foundation of China (81770236, 81500244, 81602300, 81671475, 81701427), the Project on Social Development of Jiangsu Province (BE2015614), and the National Key R&D Program of China (2016YFC1000700, 2016YFC1000703).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Figure S1.
Ratio of duplicated reads from computer simulation in the given conditions of library complexity and sequencing depth.
Additional file 2: Table S1.
Comparison of library concentration and complexity before and after cffDNA enrichment.
Additional file 3: Table S2.
Demographics of the 1404 clinical cases.
Additional file 4: Table S3.
Information of ‘no-call’ results.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, P., Liang, D., Chen, Y. et al. An enrichment method to increase cell-free fetal DNA fraction and significantly reduce false negatives and test failures for non-invasive prenatal screening: a feasibility study. J Transl Med 17, 124 (2019). https://doi.org/10.1186/s12967-019-1871-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-1871-x