
Abstract
Background
Cryptorchidism is one of the most common causes of non-obstructive azoospermia (NOA) leading to male infertility. Despite various medical approaches been utilised, many patients still suffer from infertility. MicroRNAs (miRNAs) play vital roles in the progress of spermatogenesis; however, little is known about the miRNA expression profile in the testes. Therefore, the miRNA profile was assessed in the testis of post-cryptorchidopexy patients.
Methods
Three post-cryptorchidopexy testicular tissue samples from patients aged 23, 26 and 28 years old and three testis tissues from patients with obstructive azoospermia (controls) aged 24, 25 and 36 years old were used in this study. Next-generation sequencing (NGS) was used to perform the miRNA expression profiling. Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) assays were subsequently used to confirm the results of several randomly-selected and annotated miRNAs.
Results
A series of miRNAs were found to be altered between post-cryptorchidopexy testicular tissues and control tissues, including 297 downregulated and 152 upregulated miRNAs. In the subsequent qRT-PCR assays, the expression levels of most of the selected miRNAs (9/12, P < 0.05) were consistent with the results of NGS technology. Furthermore, signal transduction, adaptive immune response and biological regulation were associated with the putative target genes of the differentially-expressed miRNAs via GO analysis. In addition, oxidative phosphorylation, Parkinson’s disease and ribosomal pathways were shown to be enriched using KEGG pathway analysis of the differentially-expressed genes.
Conclusions
This study provides a global view of the miRNAs involved in post-cryptorchidopexy testicular tissues as well as the altered expression of miRNAs compared to control tissues, thus confirming the vital role of miRNAs in cryptorchidism.
Similar content being viewed by others


Background
Male factors account for approximately 50% of infertility cases, which affect 10–15% of couples around the world [1]. Although most cases of male infertility are idiopathic with no known etiological factor, some causes (i.e. varicocele, sexual dysfunction etc.) are known [2]. Among these causes, cryptorchidism is a relatively common anomaly in the male genitalia that affects approximately 2–4% of male infants. Despite various medical approaches (i.e. surgical operations and hormone administration) being applied for years, many patients still suffer from infertility [3, 4], and little is known about the clear mechanism of spermatogenesis arrest in these patients.
Spermatogenesis is a complex process consisted of three phases including mitotic, meiotic and haploid processes [5]. These cellular events require highly regulated spatiotemporal expression of specific protein-coding genes, especailly at the post-transcriptional levels [6]. MicroRNAs (miRNAs) are a series of small noncoding RNAs that negatively regulate gene expression after transcription [7]. Research has shown that miRNAs play crucial roles in spermatogenesis [5, 6, 8,9,10,11,12,13]; for example, Lian et al. identified a series of altered miRNAs in patients with non-obstructive azoospermia (NOA) using microarray technology. These identified 154 significantly downregulated and 19 upregulated miRNAs indicated the important role of miRNAs in spermatogenesis [10]. It was reported that during mouse testicular development, up-regulation of miR-449 coincided with initiation of meiotic, and miR-449 was predominantly expressed in spermatocytes and spermatids during adult spermatogenesis. Furthermore, Cdc20b/miR-449 cluster activity was documented to be cooperatively mediated by CREMT and SOX5 during postnatal testes development [5]. Later on, Comazzetto et al. have identified the miR-34 family consisted of miR-34b/c and miR-449a/b/c as upregulated from late meiosis to sperm stage. miR-34b/c and miR-449 deletion led to sterility due to abnormal spermatozoa production with reduced motility [11]. With regards to the effects of miRNAs in cryptorchidism, Duan et al. found that miR-210, a significantly upregulated miRNA in patients with NOA, was also highly expressed in patients with cryptorchidism [12]. In addition, Moritoki et al. demonstrated that miR-135a was downregulated in unilateral undescended testes in a rat model of cryptorchidism [13].
Although some miRNAs were shown to be involved in the regulation of spermatogenesis in patients with cryptorchidism, no studies have yet investigated miRNA expression in the testis of post-cryptorchidopexy patients with NOA. Therefore this study investigated the miRNA profile in the testis of post-cryptorchidopexy patients and aimed to provide a platform to expound the mechanism of spermatogenesis arrest in post-cryptorchidopexy patients with NOA.
Methods
Ethics statement
Three patients (23, 26 and 28 years old) who underwent cryptorchidopexy but were still experiencing NOA, as well as three patients (24, 25 and 36 years old) suffering from obstructive azoospermia (OA) signed informed consent and approved the use of their tissues for research purposes. The local medical ethics committee approved this study.
Clinical specimen collection
Testes tissues were collected by testicular biopsy from all six subjects between July 2017 and January 2018 at the Reproductive Medicine Center, First Affiliated Hospital of Anhui Medical University (Hefei, Anhui, China). For post-cryptorchidopexy patients, all cases were bilateral. Case one was 23 years old and underwent the operation 1 year ago, case two was 26 years old and underwent the operation 18 years ago and case three was 28 years old and underwent the operation 12 years ago. Testes samples were frozen at − 80 °C in RNAlater (Ambion, USA) immediately after surgery. Haematoxylin and eosin (HE) staining and the Johnson score system were used to assess testicular spermatogenic function.
Construction of a smRNA library and next-generation sequencing (NGS)
Total RNA was extracted from the six samples using TRIzol (Life Technologies, USA) and was used to construct miRNA libraries using the NEBNext® Multiplex Small RNA Library Prep Set (Illumina®) according to the manufacturer’s instructions. Sequencing was performed on a Hiseq X (Illumina) using the HiSeq X Reagent Kit v2.
Data analyses and novel miRNA exploration
Data were analysed according to previously-reported methods. Known miRNAs were identified by mapping reads to miRBase (version 21.0) in Homo sapiens, whilst nonmatched reads were subsequently aligned against other noncoding RNAs within the Ensembl database [14]. The remaining nonannotated sequences were selected for alignment with the integrated human transcriptome to explore novel miRNAs. All hairpin-like structures containing unclassified smRNA reads (no less than 45 reads) were predicted using miRDeep2 [15] following the criteria described previously [16].
Bioinformatic analyses for miRNAs with differential expression patterns
The target genes of the differentially-expressed miRNAs in the two groups were predicted using TargetScan [17] and miRanda [18]. Enriched GO terms and KEGG pathway analysis was subsequently applied to predict the target genes of miRNAs with differential expression patterns in the two groups of specimens.
QRT-PCR verification for altered miRNA expression
cDNA synthesis was performed using a PrimeScript RT reagent kit following the manufacturer’s instructions (Takara, Japan). The abundance of individual miRNAs was subsequently assessed via an Applied Biosystems 7500 PCR System (Applied Biosystems) using SYBR Premix Ex Taq II (Tli RNaseH Plus, Takara) under optimised reaction conditions. The specific reverse transcription and qPCR primers for all miRNAs are listed in Additional file 1. The processes were performed in accordance with the protocols supplied by the manufacturers. Briefly, for qPCR, triplicate reactions were performed at 95 °C for 10 min, and the subsequent 40 amplification cycles were conducted at 95 °C for 15 s and 60 °C for 60 s. Meanwhile, 18S rRNA was used as an internal normalised control. Relative miRNA abundances were calculated using 2−△△Ct (threshold cycle) formula, where △Ct = CtmiRNA − Ct18S rRNA and △△Ct = (△Ctpost-cryptorchidopexy − △Ctobstructive azoospermia). The miRNA concentration differences between post-cryptorchidopexy and control tissues were analysed using unpaired t-tests. P < 0.05 indicated a statistically significant difference.
Results
Histopathological characteristics of post-cryptorchidopexy testicular tissue and control tissue
To clarify the histopathological characteristics of the post-cryptorchidopexy testicular tissue (hereafter referred to as ‘cryptorchid tissue’) and control tissue (hereafter referred to as ‘normal tissue’), HE staining and the Johnson scoring system were used to assess the function of spermatogenesis (Fig. 1). The Johnson scores were 3, 3 and 3 in cryptorchid tissues, which indicated maturation arrest, and 9, 9 and 10 in normal control tissues, which indicated normal spermatogenesis.

HE-staining of cryptorchid tissue and control tissue, which clarify the histopathological characteristics of cryptorchid tissues (a) and control tissues (b).
Comprehensive overview of whole genome smRNAs in cryptorchid and normal tissues
All smRNAs [18–32 nucleotides (nt)] acquired from cryptorchid and normal tissues were deep sequenced by NGS. A total of 19,931,698 (out of 21,212,215) and 20,243,124 (out of 21,524,351) sequence reads that aligned to the human genome sequence dataset were obtained in the cryptorchid and normal tissues, respectively. MiRNAs accounted for 85.5% and 71.19% in cryptorchid and normal tissues, respectively (Fig. 2).

Results of geneome mapping and distribution of RNAs among different categories.
The most abundant of these smRNAs in cryptorchid tissue were 21 nt in length, and these smRNAs were more abundant than the 22-nt and 23-nt RNAs which were in second and third place, respectively. However, the most abundant smRNAs in normal tissue were 22 nt in length, and these were more abundant than the 21-nt and 23-nt RNAs which were in second and third place, respectively (see Additional file 2).
Understanding the distribution pattern of miRNA genes may help to elucidate their roles, therefore the chromosomal locations of miRNA genes were evaluated. In cryptorchid tissue, most miRNA genes were located on chromosomes X, 9, 3 and 21. Similarly, in normal tissues, most miRNA genes were located on chromosome X, 15, 9 and 5 (see Additional file 3).
Features of the most abundant miRNAs in cryptorchid and normal tissues
The NGS results were used to compile a list of the 20 most abundant and known miRNAs in cryptorchid tissue and the 10 most abundant and novel miRNAs in normal tissue. In cryptorchid tissue, miR-514a-3p, miR-143-3p, miR-26a-5p, miR-99a-5p, miR-202-5p, miR-509-3-5p, miR-10b-5p, miR-508-3p, let-7 g-5p and let-7f-5p were the most abundant known miRNAs. In normal tissue, miR-514a-3p, miR-143-3p, miR-26a-5p, miR-509-3-5p, miR-99a-5p, miR-202-5p, miR-10b-5p, let-7f-5p, miR-508-3p and let-7 g-5p were the most abundant known miRNAs (Table 1). Detailed information is shown in Table 1. Of the 10 most abundant novel miRNAs, only one was different between cryptorchid and normal tissues. Detailed information is shown in Table 2.
Differential expression of miRNAs between cryptorchid and normal tissues
As described previously by Zhang et al. [16], miRNAs were considered to be significantly differentially expressed between cryptorchid and normal tissues if they were altered by at least two-fold with P < 0.05 on the t-test. The results showed that 449 miRNAs were significantly differentially expressed in cryptorchid tissue (Fig. 3). Of these, 297 were downregulated and 152 were upregulated compared to normal tissue. The 30 most downregulated and upregulated known miRNAs are listed in Tables 3 and 4, respectively.

The overview of the volcano plot generated by miRNAs profile in cryptorchid tissues and control tissues.
Validating the altered expression level of miRNAs by qRT-PCR
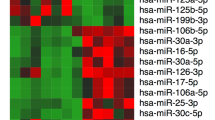
QRT-PCR was performed to validate the altered miRNA expression. Among these deregulated miRNAs, we firstly selected two well-established spermatogenesis-associated miRNAs, miR-449a and miR-34c-5p [5, 11]. Additionally, to better proving the accuracy of NGS, the other validated miRNAs were picked from the non-top 30 most deregulated known miRNAs (see Additional file 4 and Additional file 5), so that the relatively small fold changes could be validated. According to the previous studies, ten miRNAs were picked for qRT-PCR validation randomly [16, 19]. Eventually, a total of 12 differentially-expressed miRNAs (seven upregulated and five downregulated) were selected for qRT-PCR analysis. The results showed that the expression levels of most miRNAs (9/12; P < 0.05) were consistent with the results of NGS technology. Detailed information is shown in Fig. 4.

Confirmation of differentially expressed miRNAs between cryptorchid tissues and control tissues obtained by NGS using qRT-PCR. (* P <0.05)
GO enrichment analysis of differentially-expressed genes in cryptorchid and normal tissues
After predicting the target genes of differentially-expressed miRNAs in cryptorchid and normal tissues, GO enrichment analysis was conducted. The 10 most enriched GO terms, including signal transduction and adaptive immune response, are shown in Table 5.
KEGG pathway analysis of differentially-expressed genes in cryptorchid and normal tissues
After GO analysis, KEGG pathway enrichment analysis was performed. A total of five KEGG pathways were enriched, including oxidative phosphorylation, Parkinson’s disease, Ribosomal pathways, Huntington’s disease and Alzheimer’s disease. The results are presented in Table 6.
Discussion
As one of the most common congenital defects in newborn boys, cryptorchidism influences male fertility and increases the risk of testicular cancer. Reductions in seminiferous tubules and germ cells are common histological changes in cryptorchid testis [20]. Despite surgery being recommended for many patients with cryptorchidism, the success of orchidopexy depends on the timing of the procedure and the position of the testis: some may not benefit from cryptorchidopexy [21, 22]. Although research has identified some biological processes involved in spermatogenic arrest in cryptorchid testis (i.e. significant apoptotic changes in germ cells), the causative roles of genes in spermatogenic arrest or apoptosis remain unclear [23,24,25,26]. This is the first study to investigate the possible mechanisms of spermatogenic arrest in cryptorchid testes by assessing the miRNA profiles in post-cryptorchidopexy testes.
Many rodent and primate models were developed to identify altered miRNAs in cryptorchid testis. For example, Duan et al. established a mouse model of cryptorchidism and showed that miR-210 was highly expressed in cryptorchid testes compared with control testes. Moreover, they showed that this miRNA regulated spermatogenesis by inhibiting the expression of NR1D2 [12]. Moritoki et al. compared the miRNA expression profiles of unilateral undescended testes with contralateral descended testes in a rat model of cryptorchidism using microarray analysis. These authors found that only miR-135a expression was lower in unilateral undescended testes and that its target, FoxO1, played essential roles in stem cell maintenance [13]. Furthermore, Duan et al., also found that miR-210 was upregulated in human cryptorchidism, thus suggesting a vital role for miRNAs in humans [12]. In this study, 297 downregulated and 152 upregulated miRNAs were identified in post-cryptorchidopexy testicular tissue compared with normal testis tissue. However, miR-210 was not significantly altered, which may be due to the different types of human cryptorchid tissue. For example, Duan et al. used cryptorchid testis tissue obtained during the cryptorchidopexy, whilst this study used post-cryptorchidopexy testicular tissue. Some miRNA expression levels may change after the operation.
Despite the insights gained into cryptorchidism over the years, the mechanism of spermatogenesis arrest in patients with this disease remains largely elusive. Germ cell apoptosis is commonly seen at the histological level in cryptorchid testes. Yin et al. revealed that cryptorchidism induced germ cell apoptosis in an experimental mouse model via p53-dependent and p53-independent pathways [23]. Liu et al. also found that the Hsf1/Phlda1 pathway participated in primary spermatocyte apoptosis in surgery-induced cryptorchid testes of rats [27]. The expression of many apoptosis-related miRNAs was also shown to be altered in post-cryptorchidopexy testicular tissues. It was reported that miR-299-5p could modulate apoptosis through autophagy in neurons and ameliorate the cognitive capacity of APPswe/PS1dE9 mice [28]. In addition, miR-299-5p was significantly upregulated in post-cryptorchidopexy testicular tissue. Similar results were also found for miR-217, miR-206 etc. Li et al. also found that miR-217 could regulate apoptosis by targeting TNFSF11 in human podocyte cells [29]. This study also identified a significant downregulation of miR-217 in post-cryptorchidopexy testicular tissue. Similarly, miR-206 was significantly upregulated in post-cryptorchidopexy testicular tissue and was shown to promoted cell apoptosis in Legg–Calvé–Perthes disease [30].
Conclusions
In summary, miRNA expression in post-cryptorchidopexy testicular tissue was profiled using NGS and compared with that of OA men with normal spermatogenesis. Several signalling pathways that are likely to be involved in spermatogenesis arrest in these patients were addressed. The results provide an important platform for future investigations into the roles of miRNAs in the progression of cryptorchidism as well as therapeutic targets to help these patients recover fertility. However, the comprehensive modulating behaviours of genes remain unclear, therefore determining the target genes and regulatory networks of these differentially-expressed miRNAs is essential in future investigations.
Abbreviations
- HE:
-
Hematoxylin-eosin
- miRNAs:
-
MicroRNAs
- NGS:
-
Next-generation sequencing
- NOA:
-
Non-obstructive azoospermia
- OA:
-
Obstructive azoospermia
- qRT-PCR:
-
Quantitative real-time reverse transcription-polymerase chain reaction
References
Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility pathways. Nat Cell Biol. 2002;4(Suppl):s41–9.
Practice Committee of the American Society for Reproductive Medicine. Diagnostic evaluation of the infertile male: a committee opinion. Fertil Steril. 2015;103:e18–25.
Kolon TF, Herndon CD, Baker LA, Baskin LS, Baxter CG, Cheng EY, et al. Evaluation and treatment of cryptorchidism: AUA guideline. J Urol. 2014;192:337–45.
Barthold JS, Gonzalez R. The epidemiology of congenital cryptorchidism, testicular ascent and orchiopexy. J Urol. 2003;170:2396–401.
Bao J, Li D, Wang L, Wu J, Hu Y, Wang Z, et al. MicroRNA-449 and microRNA-34b/c function redundantly in murine testes by targeting E2F transcription factor-retinoblastoma protein (E2F-pRb) pathway. J Biol Chem. 2012;287:21686–98.
Bouhallier F, Allioli N, Lavial F, Chalmel F, Perrard MH, Durand P, et al. Role of miR-34c microRNA in the late steps of spermatogenesis. RNA. 2010;16:720–31.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Tang D, Huang Y, Liu W, Zhang X. Up-regulation of microRNA-210 is associated with spermatogenesis by targeting IGF2 in male infertility. Med Sci Monit. 2016;22:2905–10.
Hilz S, Modzelewski AJ, Cohen PE, Grimson A. The roles of microRNAs and siRNAs in mammalian spermatogenesis. Development. 2016;143:3061–73.
Lian J, Zhang X, Tian H, Liang N, Wang Y, Liang C, et al. Altered microRNA expression in patients with non-obstructive azoospermia. Reprod Biol Endocrinol. 2009;7:13.
Comazzetto S, Di Giacomo M, Rasmussen KD, Much C, Azzi C, Perlas E, et al. Oligoasthenoteratozoospermia and infertility in mice deficient for miR-34b/c and miR-449 loci. PLoS Genet. 2014;10:e1004597.
Duan Z, Huang H, Sun F. The functional and predictive roles of miR-210 in cryptorchidism. Sci Rep. 2016;6:32265.
Moritoki Y, Hayashi Y, Mizuno K, Kamisawa H, Nishio H, Kurokawa S, et al. Expression profiling of microRNA in cryptorchid testes: miR-135a contributes to the maintenance of spermatogonial stem cells by regulating FoxO1. J Urol. 2014;191:1174–80.
Ensembl database. http://asia.ensembl.org/index.html. Accessed 28 Sept 2017.
MiRDeep2 http://www.mdc-berlin.de/8551903/en/research/research_teams/systems_biology_of_gene_regulatory_elements/projects/miRDeep. Accessed 29 Sept 2017.
Zhang L, Wei P, Shen X, Zhang Y, Xu B, Zhou J, et al. MicroRNA expression profile in penile Cancer revealed by next-generation small RNA sequencing. PLoS One. 2015;10:e0131336.
TargetScan. http://www.targetscan.org/vert_71. Accessed 2 Oct 2017.
John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363.
Zhou Y, Wang X, Zhang Y, Zhao T, Shan Z, Teng W. Circulating microrna profile as a potential predictive biomarker for early diagnosis of spontaneous abortion in patients with subclinical hypothyroidism. Front Endocrinol (Lausanne). 2018;9:128.
Agoulnik AI, Huang Z, Ferguson L. Spermatogenesis in cryptorchidism. Methods Mol Biol. 2012;825:127–47.
Taran I, Elder JS. Results of orchiopexy for the undescended testis. World J Urol. 2006;24:231–9.
Hadziselimovic F, Thommen L, Girard J, Herzog B. The significance of postnatal gonadotropin surge for testicular development in normal and cryptorchid testes. J Urol. 1986;136:274–6.
Yin Y, DeWolf WC, Morgentaler A. Experimental cryptorchidism induces testicular germ cell apoptosis by p53-dependent and -independent pathways in mice. Biol Reprod. 1998;58:492–6.
Yin Y, Stahl BC, DeWolf WC, Morgentaler A. P53 and Fas are sequential mechanisms of testicular germ cell apoptosis. J Androl. 2002;23:64–70.
Mu X, Liu Y, Collins LL, Kim E, Chang C. The p53/retinoblastoma-mediated repression of testicular orphan receptor-2 in the rhesus monkey with cryptorchidism. J Biol Chem. 2000;275:23877–83.
Li W, Bao W, Ma J, Liu X, Xu R, Wang RA, et al. Metastasis tumor antigen 1 is involved in the resistance to heat stress-induced testicular apoptosis. FEBS Lett. 2008;582:869–73.
Liu F, Xu ZL, Qian XJ, Qiu WY, Huang H. Expression of Hsf1, Hsf2, and Phlda1 in cells undergoing cryptorchid-induced apoptosis in rat testes. Mol Reprod Dev. 2011;78:283–91.
Zhang Y, Liu C, Wang J, Li Q, Ping H, Gao S, et al. MiR-299-5p regulates apoptosis through autophagy in neurons and ameliorates cognitive capacity in APPswe/PS1dE9 mice. Sci Rep. 2016;6:24566.
Li J, Liu B, Xue H, Zhou QQ, Peng L. miR-217 is a useful diagnostic biomarker and regulates human podocyte cells apoptosis via targeting tnfsf11 in membranous nephropathy. Biomed Res Int. 2017;2017:2168767.
Luo J, Han J, Li Y, Liu Y. Downregulated SOX9 mediated by miR-206 promoted cell apoptosis in Legg-calve-Perthes disease. Oncol Lett. 2018;15:1319–24.
Acknowledgements
We thank all subjects who provided the tissues for this study.
Funding
This study was funded by the the National Natural Science Foundation of China (No. 81370749).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
DT, XH and XZ designed the study.ZH and HW performed the experiments.LZ and DP analyzed the data.DT and ZH wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all patients and this study was approved by the Ethics Review Board of The First Affiliated Hospital of Anhui Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Primers used for the quantification of representative deregulated miRNAs. (XLS 23 kb)
Additional file 2:
Length distribution of clean reads from smRNA next-generation deep sequencing. (TIF 2836 kb)
Additional file 3:
Number of smRNA sequencing tags that locate on each chromosome. (TIF 3575 kb)
Additional file 4:
A collection of all the downregulated known miRNAs detected by deep sequencing in cryptorchid tissues. (XLS 45 kb)
Additional file 5:
A collection of all the upregulated known miRNAs detected by deep sequencing in cryptorchid tissues. (XLS 35 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tang, D., Huang, Z., He, X. et al. Altered miRNA profile in testis of post-cryptorchidopexy patients with non-obstructive azoospermia. Reprod Biol Endocrinol 16, 78 (2018). https://doi.org/10.1186/s12958-018-0393-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-018-0393-3