
Abstract
Field cancerization and metastasis are the leading causes for cancer recurrence and mortality in cancer patients. The formation of primary, secondary tumors or metastasis is greatly influenced by multifaceted tumor-stroma interactions, in which stromal components of the tumor microenvironment (TME) can affect the behavior of the cancer cells. Many studies have identified cytokines and growth factors as cell signaling molecules that aid cell to cell communication. However, the functional contribution of reactive oxygen species (ROS), a family of volatile chemicals, as communication molecules are less understood. Cancer cells and various tumor-associated stromal cells produce and secrete a copious amount of ROS into the TME. Intracellular ROS modulate cell signaling cascades that aid in the acquisition of several hallmarks of cancers. Extracellular ROS help to propagate, amplify, and effectively create a mutagenic and oncogenic field which facilitate the formation of multifoci tumors and act as a springboard for metastatic tumor cells. In this review, we summarize our current knowledge of ROS as atypical paracrine signaling molecules for field cancerization and metastasis. Field cancerization and metastasis are often discussed separately; we offer a model that placed these events with ROS as the focal instigating agent in a broader “seed-soil” hypothesis.
Similar content being viewed by others

Introduction
Worldwide, one in seven deaths is due to cancer; cancer causes more deaths than Acquired Immune Deficiency Syndrome, tuberculosis, and malaria combined. Recent statistics report estimates that there will be 18.1 million new cancer cases and 9.6 million cancer deaths worldwide in 2018 [1]. Current trends also suggest that cancer will remain as one of the leading causes of death and the most important barrier to increasing life expectancy globally. Cancer-related deaths have not rocketed because of significant advances in diagnosis. Improvements and a genuine postponement of death for various cancer patients are often due to better detection methods and not to better treatments [1, 2]. However, we have made less progress with traditional therapeutic options such as chemotherapy, radiotherapy, and surgery still dominates current anti-tumor treatment methods. Emerging therapeutic modalities such as chimeric antigen receptor T-cell (CAR-T) immunotherapy approach have proven to be very effective, but only a select subset of cancers responds to the treatment [3]. Furthermore, more than 90% of cancer deaths are caused by the metastatic spread of tumor cells from the primary to distant sites [4]. Yet, our understanding of this process is limited, and there are no specific therapeutic approaches to suppress cancer metastasis. Moreover, resistance to conventional chemotherapeutics and disease relapse remain persistent clinical challenges [4]. These observations imply an incomplete understanding of the cellular and biotic heterogeneity in the tumor.
Cancer is a genetic disease resulted from both internal factors (e.g., inherited mutations, immune conditions, hormones, etc.) and external factors (e.g., environment, diet, tobacco, diet, infection, radiation, among others) [2]. These factors can affect important genes such as proto-oncogenes, tumor suppressor genes and deoxyribonucleic acid (DNA) repair genes via cellular intermediates such as reactive oxygen species (ROS) [5]. ROS are major cellular intermediates. In most studies, ROS are used as an umbrella term to describe a heterogeneous group of cellular free radicals that contain oxygen (O2) derived from various intracellular processes and extracellular sources. ROS are highly reactive to biomolecules, and they can trigger multiple biological events [6]. ROS plays a contradictory role in cancer biology. Elevated ROS levels contribute to tumorigenesis, cancer progression and spreading via the promotion and maintenance of tumorigenic cell signaling which results in tumor cell proliferation, survival, autophagy, and metastasis [7]. In Table 1, we provide a non-exhaustive list of the various common ROS and their roles in cancer.
Recently, the involvement of ROS as atypical context-dependent drivers of tumorigenesis is gaining attention [8]. On one hand, excessive ROS results in anti-tumorigenic effects via promoting cell death, inducing cell cycle arrest and senescence [9]. On the other hand, it is known that tumor cells promote their survival through enhanced ROS manipulation mechanisms, such as increased antioxidant levels or increased ROS production, to maintain the delicate balance in ROS level that supports their proliferation and survival [9, 10]. For example, autophagy can be induced by ROS to remove damaged mitochondria that contribute to oxidative stress, restoring ROS to physiological level [11]. However, the deletions of autophagy-related genes such as autophagy-related 5 (ATG5), autophagy-related 7 (ATG7) and beclin-1 (BECN1) can lead to defective autophagy. These deletions can result in the deregulated degradation of damaged mitochondria, and hence, elevated ROS production as well as oxidative stress [11].
Despite current knowledge in ROS signaling in cancer biology, the dual nature of ROS is still a huge conundrum in therapeutics targeting ROS. The inhibition or elevation of ROS levels can yield drastically different results [2, 12]. Recent discussions suggested dichotomizing the effects of ROS in cancer cells into two categories: early versus late stages. Depending on the stage of cancer progression, intracellular ROS plays a different role in cancer cell survival. At precancerous and early stages of cancer, intracellular ROS promote cancer initiation via inducing oxidative and base pair substitution mutations in pro-oncogenes such as Ras and tumor suppressor genes such as p53 [13]. Apart from inducing mutations, ROS can also modify site specific amino acids side chains which alter protein structure and functions [7]. Among the amino acids, cysteine (Cys) is more prone to oxidation by ROS due to the presence of thiol group. Cys appears to be the principal actor in redox signalling, functioning as a regulatory reversible molecular switch. As cancer progresses, the accumulation of excess intracellular ROS can trigger apoptosis, tumor cells escape apoptosis by producing high levels of intracellular antioxidants [13]. In the late stages of tumor evolution, metastatic tumors developed mechanisms that exploit ROS as a springboard for the dissemination of cancer cells. As a result, whether ROS play anti-tumor or oncogenic roles may depend on the different stages of cancer development and progression.
Many canonical pathways involved in tumor-promoting inflammation and cell proliferation have been shown to be activated by ROS. Transcription factor NF-κB plays an important role in cellular processes such as immune and inflammatory response, cellular proliferation and differentiation [14]. The canonical NF-kB pathway can be activated by oxidative stress and proinflammatory cytokines [15]. The activation of the canonical NF-κB pathway is dependent on the phosphorylation of IκB-Kinase (IKK) β, the ubiquitination-mediated degradation of NF-κB inhibitor alpha (IκBα), the translocation of NF-κB into the nucleus, resulting in the transcriptional activation of target genes [16, 17]. Studies have shown that ROS can trigger the activation of the NF-κB pathway via inducing the tyrosine phosphorylation of IκBα. IκBα, which is usually phosphorylated on serine-32 and -36 by IKK, undergoes ubiquitination and degradation for activation of the NF-κB pathway [18, 19]. Exogenous addition of H2O2 induces the phosphorylation of IκBα at tyrosine-42 as well as other tyrosine residues, resulting in the degradation of IκBα and the activation of the NF-κB pathway [17, 20].
Similarly, MAPK family which consists of ERK1/2, c-Jun N-terminal kinase (JNK), MAPK-11 and the MAPK1 pathway are important intracellular signal transduction pathways involved in cellular processes such as cell survival, cell death, growth, and differentiation [21]. Studies have demonstrated that ROS can activate the epidermal growth factor receptors (EGFR) and platelet-derived growth factor (PDGF) receptors without corresponding ligands. Thus, the activation of EGFR and PDGF can activate Ras and subsequently lead to the activation the ERK pathway [18]. Such modifications to the receptors conferred ligand-independent activation of the tyrosine kinase receptors and contribute to resistance against antibody-based therapies such as anti-EGFR (e.g. cetuximab, necitumumab) or anti-PDGF (e.g. Olaratumab). Furthermore, ROS may also activate the MAPK pathway via oxidative modification of intracellular downstream kinases such as apoptosis signal-regulating kinase 1 (ASK-1), a member of the mitogen-activated protein kinase kinase kinase (MAP3K) superfamily for JNK and MAPK-11 [22].
Another important pathway in cancer is the PI3K/PTEN pathway whereby several of the signaling mediators are redox sensitive and play important roles in field cancerization and metastasis [18]. ROS can act as signaling mediators by triggering oxidative modification of specific target molecules [23]. For example, PTEN can be modified by H2O2 via oxidation of the Cys thiol groups of phosphatases, resulting in its inactivation [7, 24].
Most studies on ROS in cancer were largely focused on the primary tumor. The roles of ROS in field cancerization and metastasis, which contribute to local and distant recurrence cancers, respectively, has been gathering attention. In the review, we will discuss the role of ROS in the tumor microenvironment (TME) in driving field cancerization and metastasis.
Origin of ROS
ROS are produced by various biochemical and physiological oxidative processes in the cell. Mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase are the two major producers of ROS [25, 26]. ROS were once viewed merely as by-products of cell metabolism, but subsequent research showed that they have many roles in normal physiology. ROS serve as an important signaling molecule participating in a variety of cellular signaling pathways such as growth factor pathways, inflammation, engagement of integrins and adhesion to the extracellular matrix [27,28,29,30].
Oxidative stress contributes to aging and many diseases such as cancer, diabetes, and obesity. Oxidative stress occurs when excessive ROS accumulate in the cell due to an imbalance of oxidative and reductive activities, resulting in cellular damage. The antioxidant defense systems include superoxide dismutase (SOD), catalase, glutathione peroxide (GPx), glutathione reductase, glutathione S-transferase (GST), and glutathione, which are important to maintain a balanced level of intracellular ROS [31]. The NF-κB pathway can influence the ROS levels by increasing the expression of SOD, GPx and GST [18]. Mitochondria is an important source of intracellular ROS, such as superoxide O2•− and H2O2 [26]. Intracellular H2O2 is formed by SOD-catalyzed dismutation from O2•− generated within the mitochondrial matrix, intermembrane space, and outer membrane [26]. Increased ROS production in cancer cells can lead to elevated SOD expression as well as the inactivation of H2O2 scavenging enzymes, producing abnormally high levels of H2O2 [11].
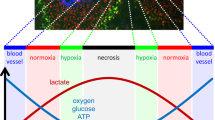
Apart from mitochondria, NADPH oxidase (NOX), an enzyme system, is a major source of extracellular ROS which mainly serves as communication molecules [2]. As integral membrane proteins, the expression of membrane-associated NOX releases O2•− into the extracellular space [32]. Furthermore, these enzymes are also internalized to form redoxosomes, extending the intracellular reach of ROS [33]. Exosomes, containing functional NOX complexes to generate ROS, can also be released from cells such as macrophages into the extracellular space and transported to distant sites [34]. An elevated intracellular ROS inevitably results in a copious amount of extracellular ROS in the TME, thus neighboring normal cells will experience oxidative stress (Fig. 1) [35].

Fate of ROS: NOX proteins are integral membrane proteins of cells and release superoxides (O2•−) as products directly into the extracellular space. Functional NOX proteins can also be internalized into forming redoxosomes, producing superoxides (O2•−) within the redoxosomes. Furthermore, from cells such as macrophages, exosomal NOX complexes can be released and being incorporated into surrounding cells via endocytosis. Besides NOX, xanthine oxidase and nitric oxide synthase proteins (both not shown) can also generate extracellular and intracellular superoxides (O2•−) respectively. Superoxides (O2•−) are relatively impermeable through the cell membrane. However, intracellular and extracellular SOD proteins catalyze the dismutation of superoxides (O2•−) into H2O2 and O2. H2O2 molecules are relatively permeable through the aquaporins of the cell membrane and hence, can travel easily from cell to cell, providing regional oxidative stress
.
ROS have critical roles in tumor pathology. A high level of intracellular ROS due to defects in ROS production or detoxification processes can transform a normal cell into a malignant cell [2]. Indeed, cancer cells have elevated levels of intracellular ROS and extracellular ROS. Cancer cells have a high level of intracellular ROS due to reasons such as increased metabolic activity and mitochondrial energetics, alterations of the electron transport chain, expression of HIF-1 due to hypoxic condition and chronic inflammation [36]. Furthermore, cancer cells also have elevated expression of membrane-associated NOX [32]. Oncogenic KRAS was reported to increase the activity of NOX enzymes on the tumor cell membrane and hence, promote extracellular ROS generation [37]. While cancer cells may be more tolerant to oxidative stress via an elevated robust antioxidant defense, the consequence may be detrimental to the TME and the adjacent normal cells. For example, cancer cells expressed membrane-associated catalase to degrade extracellular ROS as well as express membrane-associated SOD to convert the more reactive O2•− into reactive H2O2 [32, 33]. This might explain for the accumulation of oncogenic H2O2 in the TME, particularly in metastatic epithelial tumors [8, 35, 38].
Apart from cancer cells, various tumor-associated cell types also produce ROS and contribute to the oxidative microenvironment. Cancer-associated fibroblasts (CAFs) are one of the most abundant stromal cells in the TME and influence the pathology of cancer in many ways [39,40,41]. As CAFs is a cellular state rather than a cell type, CAFs are generally known as activated fibroblast in the TME with no precise molecular definition [42]. CAFs produce and are highly influenced by ROS [43]. CAFs have been shown to have an elevated level of H2O2 as compared to normal fibroblasts. The high production of intracellular and extracellular H2O2 by CAFs was due to impaired transforming growth factor beta (TGF-β) signaling [44, 45]. This impaired signaling leads to the suppression of the antioxidant enzyme GPx1 as well as the production of intracellular ROS by impaired mitochondrial function and extracellular ROS by induced NOX [46]. In addition, Caveolin-1, a negative regulator of NOX derived ROS, also increases the level of extracellular ROS production by CAFs [47]. Notably, normal fibroblasts treated with exogenous H2O2 or CAF-conditioned medium transformed into an oxidative, CAF-like state [35]. These newly transformed fibroblasts displayed elevated fibroblast activation protein (FAP) and α-smooth muscle actin (αSMA) expression levels, both of which are biomarkers of CAFs. Similar to CAFs, newly transformed fibroblasts became activated and desensitized to TGF-β. Normal fibroblasts treated with prolonged exogenous H2O2 displayed a significant increase in p65-NF-κB phosphorylation, triggering NF-κB activity. The NF-κB activation attenuated TGF-β signaling and hence, ensures the continued expression of FAP in the newly transformed fibroblasts [35].
In addition to CAFs, ROS are also released by many tumor-associated immune cells in the TME [48]. Tumor-associated macrophages (TAMs) in the TME mainly originate from blood-circulating monocytes that infiltrate into the TME and differentiate into mature pro-tumor macrophages mediated by cytokines in the TME [49,50,51]. Interestingly, the role of TAMs is a double-edged sword. As part of the immune system, macrophages are the first host cells to enter the TME and can potentially kill the cancer cells [52]. In vitro, activated macrophages showed anti-tumor activity via calreticulin binding receptors for cancer cell recognition [53]. Macrophages also display phagocytotic activity toward some damaged tumor cells [54]. However, the TME is known to have elevated levels of macrophages. During cancer initiation, cancer cells recruit macrophages via chemokines which amplify an inflammatory response. Macrophages also produce redoxosomes, i.e. exosomes containing functional NOX complexes into the TME, generating extracellular ROS and being incorporated into surrounding cells via endocytosis [34].
TAMs and CAFs are often detected close to each other, suggesting extensive communications and interactions between these two cell types [55]. The reciprocal relationship between TAMs and CAFs in the TME increases tumor malignancy, and ROS may be a key player in the interaction [56, 57]. ROS in the TME can trigger altered activation of macrophages and immunosuppression [58]. TAMs also release ROS which plays an essential role in immune alterations such as inducing apoptosis in lymphocytes [48, 59]. There was evidence indicating that the TME induces TAMs to activate immunosuppressive mechanisms via ROS production [60]. Macrophages exposed to increasing concentration of tumor fluid significantly increased intracellular ROS generation [58]. Elevated intracellular ROS corresponds to altered cellular redox homeostasis and oxidative stress [61].
Myeloid-derived suppressor cells (MDSCs) also promote cancer progression via ROS [62]. MDSCs are a heterogeneous population of cells which can suppress T cell responses and expand during inflammation and cancer [63]. These cells were first observed in patients with cancer [64,65,66]. Apart from their immunological functions, MDSCs were also reported to promote tumor angiogenesis, tumor cell invasion, and metastasis [67]. MDSCs are a unique component of the immune system which regulates the immune responses in cancer patients [63]. Indeed, up-regulation of ROS primarily by NOX is one of the major factors responsible for the immunosuppressive activity of MDSCs [68]. Granulocytic MDSCs, a subpopulation of MDSCs, were found to use ROS primarily as the mechanism of immunosuppression by close cell-cell contact with T cells [69, 70].
Although both TAMs and MDSCs were found to suppress T cell responses via different ROS mediated mechanisms, TAMs were the more potent immune suppressor [60]. MDSCs suppressed T cells via contact-independent H2O2 production and TAMs exerted their more potent immunosuppressive effects by the production of contact-dependent H2O2 [60, 69]. Certain subpopulations of MDSCs were found to be able to differentiate into immunosuppressive TAMs in the presence of tumor-derived factors or tumor-bearing hosts [69, 71, 72].
Hence, for cancer cells (the “seed”) to grow, expand and acquire more mutations to become malignant, a supportive TME (the “soil”) is required. ROS plays an essential role in creating the immunosuppressive “soil” ground for field cancerization and metastasis.
ROS in field cancerization
Field cancerization was first reported in 1953 by Slaughter et al. and is often used to describe the development of abnormal epithelia and stroma bordering a tumorigenic area [73]. This posits the presence of a regional carcinogenic signal at these foci of cellular abnormalities, and if given enough time and exposure, the carcinogenic agent will cause irreversible changes to the cells leading to oncogenic transformation and field cancerization. Indeed, field cancerization results in multifocal primary tumors in proximity with a higher chance of recurrence even after resection of the malignant tumors [74]. Presently, field cancerization basically refers to pre-malignant changes in multiple and large areas of the primary tumor, within both the epithelial cells and surrounding stromal cells [75, 76]. Despite its clinical importance, only a few cytokines such as TGF-β, macrophage inhibitory cytokine 1 and PDGF-A have been implicated as possible field carcinogens [77,78,79].
Field cancerization can be initiated and propagated in many ways, including mutagen ROS [80]. The chronic exposure of high extracellular H2O2 promoted the transformation of normal epithelial cells and fibroblasts, indicating the presence of a premalignant field defect by oxidative stress in the TME [81,82,83]. H2O2 is an ideal field effect carcinogen due to its higher cellular plasma membrane permeability and longer half-life than other ROS counterparts [7, 35]. H2O2 aggravates cancer cell aggressiveness, transform primary epithelial cells by oxidative modification of the membrane associated PTEN and Src proteins, decreasing PTEN and increasing Src activities [8, 35, 43, 84]. Normal fibroblasts treated with H2O2 transformed into an oxidative, CAF-like state. In turn, these newly converted CAF-like cells produced higher H2O2 caused by an impaired TGF-β signaling [35]. These observations indicated that stromal cells, such as CAFs, engaged redox signaling circuitries and mitogenic signalings to reinforce their reciprocal relationship with the epithelial tumor, further supports that extracellular oxidative stress might act as a field effect carcinogen [35, 43]. Thus, ROS are atypical carcinogenic signals which promote stromal-mediated field cancerization [35].
Conceivably, a single mutant cell represents the smallest origin capable of initiating field cancerization by causing neighboring normal cells to transform and to amplify an oxidative field [80]. Disseminated cancer cells can also start a new cancerized field by producing extracellular ROS. Although stromal cells such as CAFs do not transform into tumor cells themselves, alterations of stromal cells can promote field cancerization. This is because these stromal cells provide selective pressure such as oxidative stress in the field for particular newly mutated cells or disseminated cancer cells over the existing normal cells [80]. This provides the “soil” for newly mutated cells to acquire more mutations and progress towards cancer as well as newly migrated cancer cells to grow and survive in the new environment [80]. ROS in the new environment enact field cancerization by promoting oxidative stress, causing pre-malignant transformation of the surrounding cells as well as a series of immunosuppressive responses [80]. These findings suggest that the epithelial and stromal cells bathed in an oxidative milieu, continuously experienced oxidative stress that modulates their functions (Fig. 2) [35, 61].

ROS in the TME: ROS can directly or indirectly modulate the functions of many cell types in the TME. ROS are able to transform normal epithelial cells and normal fibroblasts into malignant cells and CAFs respectively. ROS produced by cancer cells, TAMs and MDSCs can have an immunosuppressive effect on T-cells and B-cells. ROS can accumulate in the extracellular matrix, causing neighboring normal cells to acquire malignant phenotypes. Migration of the ROS-producing cells (cancer cells, CAFs, TAMs, MDSCs) to distant tissue or organ can start a new cancer field, transforming neighboring normal cells into cancer supporting cells or new malignant cells. This expanding field provides the appropriate “soil” for the survival and growth of newly-disseminated cancer cells or newly-transformed cells (the “seed”)
Altogether, these results confirm the presence of stromal-mediated field cancerization, whereby the influences of stromal oxidative stress can be propagated and amplified, and effectively create a mutagenic or oncogenic field promoting multifocal tumor formations [35]. This finding features the mesenchymal-mesenchymal and epithelial-mesenchymal communications in the propagation of field effect and the creation of a TME niche [43].
ROS in cancer metastasis
Metastasis is the spread of the primary tumor cells to distant organs, and this process is considered the main cause of cancer morbidity and mortality [4]. Once metastasis occurs, surgical excision of the primary tumor no longer guarantees disease-free survival, and the probability of cancer relapse in distal organs increases significantly. Metastasis is a complex, multi-step process beginning with cancer cells in the primary tumor undergoing EMT [85, 86]. This leads to an invasive tumor epithelial phenotype characterized by detachment from and degradation of the basement membrane [87]. Eventually, the invasive cancer cells gain access to local vasculature and/or lymphatics, intravasate and enter the systemic circulation. In the absence of basement membrane attachment, circulating cancer cells circumvent anoikis and evade the immune surveillance until they arrive at a secondary site where they extravasate and colonize distal organs [88]. Although most cancer deaths are the result of metastases, cancer research has mainly focused on the primary tumor.
Metastasis begins with EMT, which is a transdifferentiation program whereby epithelial cancer cells lose cell-cell adhesion and concomitantly acquire mesenchymal features of migration and invasion [86]. Several pieces of evidence have established a strong connection between EMT of epithelial cancer cells and ROS. TGF-β1 is well-established as one of the more prominent players of the induction of EMT [89]. TGF-β1 regulates urokinase-type plasminogen activator (uPA) and MMP9 to facilitate cell migration and invasion via the activation of NF-κB through the Rac1-NOXs-ROS-dependent mechanism [90]. Similarly, ROS also plays a crucial role in the regulation of EMT via the non-canonical TGF-β1-TGF-β-activated kinase 1 (TAK1) pathway. The increase in integrin:Rac-induced ROS by TAK1 deficiency results in a cascade of signals leading to accelerated EMT. Consistently, the expression of TAK1 was reduced in invasive squamous cell carcinoma (SCC), an observation absent from benign SCCs [91]. In a recent study, Matsuno and colleagues showed that ROS regulates EMT via the activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), increasing Notch signaling which ultimately increases EMT [92]. It is known that exogenous ROS by sources such as ionizing radiation also results in the induction of TGF-β1 [93]. Taken together, increasing evidence revealed a multifaceted role of ROS in EMT. The fact that ROS is involved in several pathways that directly link to many critical EMT-inducing pathways underscores its importance and the crucial role of ROS in EMT.
Circulating cancer cells acquire anoikis resistance, where it loses its dependence on integrin-mediated extracellular matrix contact for survival and growth [38]. Many studies have shown that ROS is indeed one of the key players in anoikis sensitivity. The metastasis-associated gene, angiopoietin-like 4 (ANGPTL4) has been shown to be a key player. Via an outside-in signaling mechanism, ANGPTL4 protein engages with integrin to stimulate the production of ROS, which subsequently activates PI3K/Akt and ERK to confer anoikis resistance to tumor cells [38]. In a recent study, anoikis resistance in gastric cancer cells was attributed to an increase in NOX4-induced ROS generation [94]. The increase in ROS levels by NOX4 upregulates EGFR, which is a growth factor involved in cell survival and anoikis inhibition [95]. Similarly, another study also revealed that EGFR is directly associated with increased cell survival in the absence of extracellular matrix [96]. Indeed, intracellular ROS plays an integral role such as in the regulation of growth factors to bring about anoikis resistance of cancer cells which is an important step in metastasis.
In the final stage of successful metastasis, circulating cancer cells will extravasate and colonize the new secondary tumor site due to its predisposed microenvironment [97]. It has been revealed that interactions from primary tumor sites are able to set up a pre-metastatic niche in the secondary tumor site, and this determines the survival of disseminated tumor cell at the new site. The secondary TME also determines the outcome of the disseminated cancer cell, whether it thrives or remain dormant [98]. Increasing evidence has shown that ROS play a role in creating a ‘soil’ in distal organs, setting up a supportive tumor environment for disseminated cancer cells. One of such ways is through the cellular disposal of miR-23b via exosomes. miR-23b is a microRNA that is negatively involved in tumorigenesis through the regulation ROS [99]. The study also suggested that the transfer of exosome containing miR-23b from bone marrow mesenchymal stem cells, a common secondary tumor site in breast cancer, might enable a metastatic niche that promoted breast cancer cell dormancy, an observation that was contingent with breast cancer recurrence [100]. The accumulation of MDSCs from haemopoietic cells in the microenvironment of metastatic niches causes the increase in production of ROS that suppresses cytotoxic CD8+ T-cells activity, which promoted the disseminated cancer cell’s survival in the secondary tumor site [70, 101].
The involvement of ROS in various steps of metastasis makes it an integral player in the metastasis of tumors. The finding is important in guiding the way future clinical trials may be conducted as well as the development of redox-therapies that target the metastasis.
Conclusion and perspectives
While field cancerization and metastasis are often discussed separately, these two phenomena may be analogously represented by a growing tree that eventually sprouts branches and develops a wide canopy. At its roots, a cancerized field fuels the acquisition of mutations or transcriptome changes, i.e., the “trunk” to promote growth. This co-evolution of tumor-stroma drives tumor cell clonal selection. Hence, some branches and offshoots begin to appear, i.e., intratumor heterogeneity. ROS helps to propagate, amplify, and effectively create a mutagenic and oncogenic field will facilitate the formation of multifoci tumors and act as a springboard for metastatic tumor cells. However, not all new profile change and mutations confer a selective advantage, and therefore some branches do not fully develop. Over time, tumor cells with the appropriate profile of metastatic “driver” genes within the cancerized field become aggressive and gain the capacity to invade, intravasate, evade the immune system and metastasize. The seed-soil concept by Paget becomes relevant in determining the survival of this disseminated tumor cells [97]. Again, ROS produced by exosomes could assist to interrogate and corrupt the distant soil for more effective colonization of the disseminated cancer cells. It is attempting to speculate that metastatic dormancy, where a disseminated tumor cell remains in a quiescent state at a remote organ while waiting for appropriate environmental conditions to begin proliferation again, may be partly attributed to poorly-prepared soil. Upon engraftment within a suitable secondary site, the metastasized tumor cell may once again enact field cancerization to corrupt its new microenvironment. ROS play important roles during field cancerization and metastasis, but many events remained relatively understudied. The scarcity of mouse models to monitor the production of volatile ROS by the tumor and to identify the cells affected by ROS in vivo remains a bottleneck to our understanding.
Abbreviations
- Akt:
-
RAC-alpha serine/threonine-protein kinase
- ANGPTL4:
-
Angiopoietin-like 4
- ASK-1:
-
Apoptosis signal-regulating kinase 1
- ATG5:
-
Autophagy-related 5
- ATG7:
-
Autophagy-related 7
- BECN1:
-
Beclin-1
- CAF:
-
Cancer-associated fibroblast
- CAR-T:
-
Chimeric antigen receptor T-cell
- Cys:
-
Cysteine
- DNA:
-
Deoxyribonucleic acid
- EGFR:
-
Epidermal growth factor receptors
- EMT:
-
Epithelial-mesenchymal transition
- ERK:
-
Extracellular signal-related kinases
- FAP:
-
Fibroblast activation protein
- GPx:
-
Glutathione peroxide
- GST:
-
Glutathione S-transferase
- HIF:
-
Hypoxia-inducible factor
- IKK:
-
IκB-Kinase
- IκBα:
-
NF-κB inhibitor alpha
- JNK:
-
c-Jun N-terminal kinase
- MAP3K:
-
mitogen-activated protein kinase kinase kinase
- MAPK:
-
Mitogen-activated protein kinase
- MDSC:
-
Myeloid-derived suppressor cell
- MMP:
-
Matrix metalloproteinase
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NOX:
-
NADPH oxidase
- Nrf2:
-
Nuclear factor (erythroid-derived 2)-like 2
- PDGF:
-
Platelet-derived growth factor
- PI3K:
-
Phosphoinositide 3-kinases
- PTEN:
-
Phosphatase and tensin homolog
- ROS:
-
Reactive oxygen species
- SCC:
-
Squamous cell carcinoma
- SOD:
-
Superoxide dismutase
- TAK1:
-
TGF-β-activated kinase 1
- TAM:
-
Tumor-associated macrophage
- TGF-β:
-
Transforming growth factor beta
- TME:
-
Tumor microenvironment
- uPA:
-
Urokinase-type plasminogen activator
- αSMA:
-
α-smooth muscle actin
References
Bray, F., et al., Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 2018.
Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: role of antioxidative nutraceuticals. Cancer Lett. 2017;387:95–105.
Androulla MN, Lefkothea PC. CAR T-cell therapy: a new era in Cancer immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5–18.
Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18(1–2):43.
Sadikovic B, et al. Cause and consequences of genetic and epigenetic alterations in human cancer. Current genomics. 2008;9(6):394–408.
Murphy MP, et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011;13(4):361–6.
Kumari S, Badana AK, Malla R. Reactive oxygen species: a key constituent in cancer survival. Biomark Insights. 2018;13:1177271918755391.
Arcucci A, et al. Cancer: an oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. Biomed Res Int. 2016:2016.
Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–6.
Galadari S, et al. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144–64.
Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018(80);50–64.
Raza MH, et al. ROS-modulated therapeutic approaches in cancer treatment. J Cancer Res Clin Oncol. 2017;143(9):1789–809.
Assi M. The differential role of reactive oxygen species in early and late stages of cancer. Am J Phys Regul Integr Comp Phys. 2018.
Park M, Hong J. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells. 2016;5(2):15.
Pramanik K, et al. Advancement of NF-κB signaling pathway: a novel target in pancreatic Cancer. Int J Mol Sci. 2018;19(12):3890.
Hinz M, Scheidereit C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014;15(1):46–61.
Liu T, et al. NF-κB signaling in inflammation. Signal Transduction Targeted Ther. 2017;2:17023.
Zhang J, et al. ROS and ROS-mediated cellular signaling. Oxidative Med Cell Longev. 2016:2016.
Lee Y, et al. Transient exposure to hydrogen peroxide inhibits the ubiquitination of phosphorylated IκBα in TNFα-stimulated HEK293 cells. Exp Mol Med. 2012;44(8):513.
Liu C, et al. The RCAN1 inhibits NF-κB and suppresses lymphoma growth in mice. Cell Death Dis. 2015;6(10):e1929.
Manna PR, Stocco DM. The role of specific mitogen-activated protein kinase signaling cascades in the regulation of steroidogenesis. J Signal Transduction. 2011;2011.
Son Y, et al. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduction. 2011:2011.
Corcoran A, Cotter TG. Redox regulation of protein kinases. FEBS J. 2013;280(9):1944–65.
Truong TH, Carroll KS. Redox regulation of protein kinases. Crit Rev Biochem Mol Biol. 2013;48(4):332–56.
Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313.
Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13.
Sundaresan M, et al. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–9.
Leto TL, Geiszt M. Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal. 2006;8(9–10):1549–61.
Werner E, Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on rho GTPases. J Cell Biol. 2002;158(2):357–68.
Chiarugi P, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161(5):933–44.
Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113(7):4633–79.
Bauer G. Targeting extracellular ROS signaling of tumor cells. Anticancer Res. 2014;34(4):1467–82.
Nordzieke D, Medraño-Fernandez I. The plasma membrane: a platform for intra-and intercellular redox signaling. Antioxidants. 2018;7(11):168.
Hervera A, et al. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol. 2018;20(3):307.
Chan JSK, et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell Death Dis. 2017;8(1):e2562.
Tong L, et al. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015;367(1):18–25.
Suh Y, Lee S-J. KRAS-driven ROS promote malignant transformation. Mol Cell Oncol. 2015;2(1):e968059.
Zhu P, et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2−: H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19(3):401–15.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582.
Shiga K, et al. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers. 2015;7(4):2443–58.
Spaw M, Anant S, Thomas SM. Stromal contributions to the carcinogenic process. Mol Carcinog. 2017;56(4):1199–213.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30(9):1002–19.
Liao Z, et al. Cancer-associated fibroblasts in tumor microenvironment - accomplices in tumor malignancy. Cell Immunol. 2018;S0008-8749(17):30222–8.
Avagliano, A., et al., Metabolic reprogramming of Cancer associated fibroblasts: the slavery of stromal fibroblasts. Biomed Res Int, 2018. 2018.
Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1(4):482.
Liu R-M, Desai LP. Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–77.
Chen F, et al. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. 2014;73:201–13.
Karin, M., NF-κB as a critical link between inflammation and cancer. Cold Spring Harbor perspectives in biology, 2009: p. a000141.
Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;2:73.
Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;1:1.
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–65.
Munn LL. Cancer and inflammation. Wiley Interdiscip Rev Syst Biol Med. 2017;9(2):e1370.
Feng, M., et al., Macrophages eat cancer cells using their own calreticulin as a guide : roles of TLR and Btk. Proc Natl Acad Sci U S A, 2015(7): p. 2145.
Nao e, et al. Recognition of damage-associated, nucleic acid-related molecular patterns during inflammation and vaccination. Front Cell Infect Microbiol. 2013;2:2013.
Herrera M, et al. Cancer-associated fibroblast and M 2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;104(4):437–44.
Hashimoto O, et al. Collaboration of cancer-associated fibroblasts and tumour-associated macrophages for neuroblastoma development. J Pathol. 2016;240(2):211–23.
Comito G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33(19):2423.
Ghosh S, et al. Reactive oxygen species in the tumor niche triggers altered activation of macrophages and immunosuppression: role of fluoxetine. Cell Signal. 2015;27(7):1398–412.
Enukidze, M., et al., Cell death in Jurkat cells induced by oxygen/nitrogen stress. Georgian Med News, 2009(167): p. 109–113.
Hamilton MJ, et al. Macrophages are more potent immune suppressors ex vivo than immature myeloid-derived suppressor cells induced by metastatic murine mammary carcinomas. J Immunol. 2014;192(1):512–22.
Mukherjee S, et al. Pomegranate reverses methotrexate-induced oxidative stress and apoptosis in hepatocytes by modulating Nrf2-NF-κB pathways. J Nutr Biochem. 2013;24(12):2040–50.
OuYang L-Y, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. 2015;13(1):47.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162.
Young MR, Newby M, Wepsic HT. Hematopoiesis and suppressor bone marrow cells in mice bearing large metastatic Lewis lung carcinoma tumors. Cancer Res. 1987;47(1):100–5.
Buessow SC, Paul RD, Lopez DM. Influence of mammary tumor progression on phenotype and function of spleen and in situ lymphocytes in mice. J Natl Cancer Inst. 1984;73(1):249–55.
Seung LP, et al. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci. 1995;92(14):6254–8.
Murdoch C, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618.
Corzo CA, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40(11):2969–75.
Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13(7):828.
Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174(8):4880–91.
Narita Y, et al. Potential differentiation of tumor bearing mouse CD11b+ gr-1+ immature myeloid cells into both suppressor macrophages and immunostimulatory dendritic cells. Biomed Res. 2009;30(1):7–15.
Slaughter DP, Southwick HW, Smejkal W. “Field cancerization” in oral stratified squamous epithelium. Clinical implications of multicentric origin. Cancer. 1953;6(5):963–8.
Dotto GP. Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J Clin Invest. 2014;124(4):1446–53.
Seton-Rogers S. Tumour microenvironment: field effect. Nat Rev Cancer. 2012;12(8):508.
Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149(6):1179–81.
Jones AC, et al. Prostate field cancerization: Deregulated expression of macrophage inhibitory cytokine 1 (MIC-1) and platelet derived growth factor A (PDGF-A) in tumor adjacent tissue. PLoS One. 2015;10(3):e0119314.
Saito A, et al. The role of TGF-β signaling in lung Cancer associated with idiopathic pulmonary fibrosis. Int J Mol Sci. 2018;19(11):3611.
Hu B, et al. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell. 2012;149(6):1207–20.
Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer. 2018;18(1):19.
Jena N. DNA damage by reactive species: mechanisms, mutation and repair. J Biosci. 2012;37(3):503–17.
Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013;5(2):a012559.
Kang M, et al. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2013;3(1):e249.
Li X, et al. A CCL2/ROS autoregulation loop is critical for cancer-associated fibroblasts-enhanced tumor growth of oral squamous cell carcinoma. Carcinogenesis. 2014;35(6):1362–70.
Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015;36:13–22.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med. 2013;91(4):411–29.
Saitoh M. Regulation of EMT by TGF-β Signaling in Cancer Cells, in Regulation of Signal Transduction in Human Cell Research: Springer; 2018. p. 71–84.
Tobar N, Villar V, Santibanez JF. ROS-NFκΒ mediates TGF-β1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem. 2010;340(1–2):195–202.
Lam C, et al. Loss of TAK1 increases cell traction force in a ROS-dependent manner to drive epithelial–mesenchymal transition of cancer cells. Cell Death Dis. 2013;4(10):e848.
Matsuno, Y., et al., ROS-Nrf2 Pathway Mediates the Development of TGF-b1-Induced Epithelial-Mesenchymal Transition Through the Interaction with Notch Signaling, in B61. EPITHELIAL CELL BIOLOGY IN RESPIRATORY DISEASE. 2018, American Thoracic Society. p. A3805-A3805.
Robertson IB, Rifkin DB. Unchaining the beast; insights from structural and evolutionary studies on TGFβ secretion, sequestration, and activation. Cytokine Growth Factor Rev. 2013;24(4):355–72.
Du S, et al. NADPH oxidase 4 regulates anoikis resistance of gastric cancer cells through the generation of reactive oxygen species and the induction of EGFR. Cell Death Dis. 2018;9(10):948.
Kim H, et al. Regulation of anoikis resistance by NADPH oxidase 4 and epidermal growth factor receptor. Br J Cancer. 2017;116(3):370.
Reginato MJ, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5(8):733.
Paget G. Remarks on a case of alternate partial anaesthesia. Br Med J. 1889;1(1462):1.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239.
Donadelli M, et al. Regulation of miR-23b expression and its dual role on ROS production and tumour development. Cancer Lett. 2014;349(2):107–13.
Ono, M., et al., Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal, 2014. 7(332): p. ra63.
Sceneay J, et al. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology. 2013;2(1):e22355.
Ježek J, Cooper K, Strich R. Reactive oxygen species and mitochondrial dynamics: the yin and Yang of mitochondrial dysfunction and Cancer progression. Antioxidants. 2018;7(1):13.
Reczek CR, Chandel NS. The two faces of reactive oxygen species in cancer. Annu Rev Cancer Biol. 2017;1:79–98.
Wang Y, et al. Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. 2018;217(6):1915–28.
Barrera G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN oncology. 2012;2012.
Gurer-Orhan H, et al. The role of oxidative stress modulators in breast cancer. Curr Med Chem. 2018;25(33):4084–101.
Acknowledgements
We thank Dr. Jeremy Soon Kiat CHAN for his perspective and discussion on field cancerization and metastasis. We also thank Dr. Hong Sheng CHENG and Miss Jeannie Xue Ting LEE for their comments and help for this paper.
Funding
Authors’ works described in this article were supported by grants from Ministry of Education, Singapore (2014-T2–1-012 and 2017-T1–002–103) to NST, and ZL is a recipient of the NTU Research Scholarship and NTU-KI Joint PhD Scholarship from the School of Biological Sciences, Nanyang Technological University Singapore.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
NST conceptualized the discussions. ZL and DC wrote the manuscript under the supervision of NST. NST edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liao, Z., Chua, D. & Tan, N.S. Reactive oxygen species: a volatile driver of field cancerization and metastasis. Mol Cancer 18, 65 (2019). https://doi.org/10.1186/s12943-019-0961-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-0961-y