
Abstract
Background
Familial hypocalciuric hypercalcemia (FHH) is a rare autosomal dominant disease, which requires differential diagnosis from relatively common primary hyperparathyroidism (PHPT) in order to avoid unnecessary surgery.
Case presentation
A 16-year-old female had been followed by the department of psychosomatic medicine at our institution. Throughout the follow-up period, her plasma calcium levels were high, plasma Pi levels were relatively low, and plasma intact PTH was relatively high. She was referred to our department to determine the cause of her hypercalcemia. Her 24 h urinary calcium excretion was as low as 100 mg/day, and calcium creatinine clearance ratio was below 0.01. Moreover, she had a family history of hypercalcemia (proband, her brother, and her father). The genetic testing for her family revealed that she, her brother, and her father were definitively diagnosed with FHH type 1 due to the heterozygous calcium-sensing receptor mutation (NM_00388:4:c.164C > T:p.Pro55Leu).
Conclusion
We experienced a 16-year-old female with FHH, in whom genetic testing identified the heterozygous calcium-sensing receptor mutation (NM_00388:4:c.164C > T:p.Pro55Leu) as pathogenic, permitting a definitive diagnosis of FHH type 1. The genetic testing for calcium sensing receptor is beneficial to distinguish asymptomatic primary hyperparathyroidism from FHH.
Similar content being viewed by others


Background
Hypercalcemia is a condition causing disorders in multiple organs, including bone, brain, and heart [1]. The causes of hypercalcemia include hyperparathyroidism, cancer, tuberculosis, immobility, and inappropriate vitamin D supplementation. Among these, primary hyperparathyroidism (PHPT) is a relatively common disease (incidence rate;1 in 1,000) and often requires parathyroid gland resection [2]. Familial hypocalciuric hypercalcemia (FHH) is a rare autosomal dominant disease (incidence rate; 1 in 78,000) that requires, in general, no treatment [2, 3]. Thus, differential diagnosis of FHH from PHPT is critical to avoid unnecessary surgery.
Urinary calcium excretion and calcium creatinine clearance ratio (CCCR) levels are widely used for differential diagnosis of FHH and PHPT; even so, it is sometimes difficult to distinguish the two diseases [2,3,4,5,6]. Approximately 80% of FHH patients demonstrate urinary calcium excretion < 100 mg/day and CCCR < 0.01, while ~ 20% show CCCR 0.01–0.02 [6]. In contrast, PHPT patients usually have CCCR > 0.02, while ~ 20%, who have concomitant vitamin D deficiency show CCCR < 0.01 [6]. It is therefore difficult to distinguish FHH from PHPT based only on CCCR values. Thus, genetic testing becomes critical for the positive diagnosis of FHH.
To date, FHH-related mutations have been identified in genes related to the calcium sensing system that regulates calcium homeostasis [2]. The calcium-sensing receptor, CASR, is encoded by the CASR gene and belongs to a G protein-coupled receptor family that regulates parathyroid hormone (PTH) secretion, vitamin D synthesis, and calcium absorption and resorption [2, 5]. G-protein α11 (GNA11) is encoded by the GNA11 gene and mediates CaSR signaling [2, 5]. Adaptor-related protein complex 2 (AP-2) is a membrane-associated heterotetramer complex and composed of two large chain, medium chain, and a small chain. The adaptor-related protein complex 2 sigma one subunit encoded by the AP2S1 gene binds to CASR and facilitates CaSR internalization [2, 5]. Loss of function mutations in the CASR, GNA11, or AP2S1 genes cause FHH type 1 (FHH1), FHH type 2 (FHH2), and FHH type 3 (FHH3), respectively [1, 4]. Approximately 60% of FHH patients have mutations in the CASR gene [2].
Here we report a case of FHH1 in which genetic testing identified the heterozygous CASR mutation p.Pro55Leu, which permitted the definitive diagnosis of the disease. In this case with no symptom, family history and genetic test for CASR was beneficial for the diagnosis of FHH type1.
Case presentation
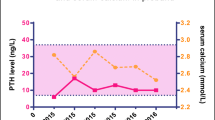
A 16-year-old female had been followed by the department of psychosomatic medicine at our institution for borderline personality disorder. Throughout the follow-up period, her plasma calcium levels were high, plasma Pi levels were relatively low, and plasma intact PTH (PTHi) was relatively high (Fig. 1).

Changes in plasma calcium, inorganic phosphate and intact parathyroid hormone in the current case. Square, plasma calcium; triangle, inorganic phosphate (Pi); circle, intact PTH (PTHi)
Her height and body weight were 154 cm and 53. 5 kg, respectively. Although she was asymptomatic, she was referred to our department to determine the cause of her hypercalcemia on June. Upon referral, her plasma calcium, inorganic phosphate (Pi) and PTHi were 11.5 (normal range; 8.8–10.1) mg/dL, 3.1 (normal range; 2.7–4.6) mg/dL, and 51 (normal range; 10–65) pg/mL, respectively (Table 1 and Fig. 1). Her 1,25-OH vitamin D (1,25-OHD) was elevated (59.7 [normal range; 20.0–60.0]) while 25-OH vitamin D (25-OHD) was low (9 [normal range; < 25]) (Table 1). Thyroid echography and 99mTc-MIBI scintigraphy revealed no abnormalities in the parathyroid glands. Her 24 h urinary calcium excretion was as low as 100 (normal range; 100–300) mg/day, and CCCR was below 0.01 (1st measurement, 0.0053 and 2nd measurement, 0.0059) (normal range; > 0.01) (Table 1). Her 24 h urinary Pi excretion was 0.58 (normal range; 0.5–1.0) g/day, and tubular reabsorption of phosphate was 89.9% (normal range; 80–94) (Table 1). Her TmP/GFR was almost same as plasma Pi levels, which were consistent with her normal renal function (Table 1).
Since her brother (II-1) and father (I-1) had hypercalcemia (Fig. 2A), we proposed that she receive a genetic test to diagnose with FHH and she and her family agreed with our proposal. Whole exon sequencing was performed by fragmentation of DNA, enrichment of exonic regions using capture-based hybridization, PCR amplification, and rapid sequencing using a next-generation sequencer in Kazusa DNA laboratory. Exon sequencing of hereditary hypercalcemia-related genes (e.g., MEN1, CDKN1B, RET, CASR, GNA11, AP2S1, CDC73, GCM2) was performed and a pathogenic CASR mutation (CASR: NM_000388.4: exon2: c.164C > T: p. Pro55Leu) was noted in her father (I-1), brother (II-1), and the proband (II-3) (Fig. 2B), but no mutation in GNA11 and AP2S1 was identified. Based on the American College of Medical Genetics and Genomics Standards and Guidelines, the present variant was classified as pathogenic due to PS1 + PS3 + PM1 + PM2 [7]. She was then positively diagnosed as FHH1. As she was diagnosed with FHH, we followed up her with no medication. She and her family were relieved to hear that she needed no surgical treatment. Her plasma Ca2+ and Pi levels were almost unchanged for 18 months (Fig. 1).

A family tree of the patient and the results of genetic testing in the patient and her family. A Squares, circles, and arrows indicate males, females, and proband, respectively. Family members, including I-1, II-1, II-3, had a history of hypercalcemia. d. Diseased. M and N, CASR wild type and p. Pro55Leu mutation alleles, respectively. Roman numerals on the left of the pedigrees indicate generation number, and the numbers below the symbols indicate the subject's number within each pedigree. The arrow shows the proband. B Sequence analysis of this family. M and N, CASR wild type and p. Pro55Leu mutation alleles, respectively. The arrow shows this mutation site (c.164). The half-filled symbols indicate the individuals with FHH
Discussion and conclusions
We report here a case of hypercalcemia with family history and low CCCR with hypovitaminosis D, in which genetic testing permitted definitive diagnosis of FHH1.
FHH is a very rare benign inherited condition that typically does not require parathyroidectomy. Patients with FHH usually have no symptoms and are often diagnosed by chance during routine blood examination. Weakness, fatigue, issues with concentration, constipation, polyuria, headache, and polydipsia have been reported by some people with FHH. The calcimimetic drugs are sometimes effective to improve hypercalcemic symptoms such as muscle aches, anorexia, polydipsia and constipation [2]. Rarely, people with this disorder experience pancreatitis or a chondrocalcinosis [8]. Although she had no past-history of pancreatitis and bone fractures, we may follow up her risk of pancreatitis due to hypercalcemia through her life [8].
In the current study, a heterozygous CASR mutation p.Pro55Leu was found to be pathogenic in the patient’s father (I-1), brother (II-1) and the proband (II-3). This mutation has previously been found in several individuals with FHH including one Japanese [2, 8,9,10,11,12,13,14,15,16]; it was classified as pathogenic in the ClinVar database (Variation ID: 279,731). The mutation is localized in the N-terminal extracellular domain (ECD) of CaSR, where ligand binding occurs; it causes a rightward shift of the PTH dose–response curve in response to extracellular calcium [11, 16]. Consistent with available information regarding the mutation, the patient showed relatively high PTHi despite the high plasma calcium level (Table 1 and Fig. 1).
In conclusion, we report a case of FHH1 due to CASR p.Pro55Leu mutation, in which only genetic testing allowed definitive diagnosis of FHH1 and thus avoided unnecessary surgery. The patient and family commented that they were very relieved to have found the cause of the disease. To diagnose with FHH by a genetic test is important to avoid unnecessary surgical treatment.
Availability of data and materials
Clinical data from the corresponding author will be available upon request.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- FHH:
-
Familial hypocalciuric hypercalcemia
- PHPT:
-
Primary hyperparathyroidism
- CCCR:
-
Calcium creatinine clearance ratio
- CASR:
-
Calcium sensing receptor
References
Turner JJO. Hypercalcaemia - presentation and management. Clin Med (Lond). 2017;17(3):270–3.
Lee JY, Shoback DM. Familial hypocalciuric hypercalcemia and related disorders. Best Pract Res Clin Endocrinol Metab. 2018;32(5):609–19.
Hinnie J, Bell E, McKillop E, Gallacher S. The prevalence of familial hypocalciuric hypercalcemia. Calcif Tissue Int. 2001;68(4):216–8.
Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Brixen K, et al. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin Endocrinol (Oxf). 2008;69(5):713–20.
Vargas-Poussou R, Mansour-Hendili L, Baron S, Bertocchio JP, Travers C, Simian C, et al. Familial Hypocalciuric Hypercalcemia Types 1 and 3 and Primary Hyperparathyroidism: Similarities and Differences. J Clin Endocrinol Metab. 2016;101(5):2185–95.
Heath H 3rd. Familial benign (hypocalciuric) hypercalcemia. A troublesome mimic of mild primary hyperparathyroidism. Endocrinol Metab Clin North Am. 1989;18(3):723–40.
Richards S, Aziz N, Bale S, Bick D, Das S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Marx SJ. Calcimimetic use in familial hypocalciuric hypercalcemia-a perspective in endocrinology. J Clin Endocrinol Metab. 2017;102(11):3933–6.
Hendy GN, D’Souza-Li L, Yang B, Canaff L, Cole DE. Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2000;16(4):281–96.
Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, et al. Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum Mol Genet. 2012;21(12):2768–78.
Pearce SH, Bai M, Quinn SJ, Kifor O, Brown EM, Thakker RV. Functional characterization of calcium-sensing receptor mutations expressed in human embryonic kidney cells. J Clin Invest. 1996;98(8):1860–6.
Guarnieri V, Canaff L, Yun FH, Scillitani A, Battista C, et al. Calcium-sensing receptor (CASR) mutations in hypercalcemic states: studies from a single endocrine clinic over three years. J Clin Endocrinol Metab. 2010;95(4):1819–29.
Pearce SH, Trump D, Wooding C, Besser GM, Chew SL, et al. Calcium-sensing receptor mutations in familial benign hypercalcemia and neonatal hyperparathyroidism. J Clin Invest. 1995;96(6):2683–92.
Fukumoto S, Chikatsu N, Okazaki R, Takeuchi Y, Tamura Y, et al. Inactivating mutations of calcium-sensing receptor results in parathyroid lipohyperplasia. Diagn Mol Pathol. 2001;10(4):242–7.
Cetani F, Pardi E, Borsari S, Tonacchera M, Morabito E, et al. Two Italian kindreds with familial hypocalciuric hypercalcaemia caused by loss-of-function mutations in the calcium-sensing receptor (CaR) gene: functional characterization of a novel CaR missense mutation. Clin Endocrinol (Oxf). 2003;58(2):199–206.
Walton RJ, Bijvoet OL. Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2(7929):309–10.
Acknowledgements
We thank all of the patients included in this study. The authors also thank H. Tsuchida, J. Kawada, and M. Kato for their technical assistance and M. Yato, Y. Ogiso, and M. Nozu for their secretarial assistance. The genetic testing was carried out at Kazusa DNA laboratory (Chiba, Japan).
Funding
This work was supported by grants from the Japan Society for the Promotion of Sciences [20K11645 (K.I.), 20K19673 (L.Y.), 21K19504 (D.Y.)]. The funders had no role on the design of study and collection, analysis, and interpretation of data and in writing the manuscript in this study.
Author information
Authors and Affiliations
Contributions
AS, KI, and DY contributed to the analysis, collection, and interpretation of data and writing of the manuscript. KT, YL, SK, SK-O, TS, TI, YT, MM, KT, TH, TS, YH, MY, YS and AS contributed to the analysis, collection, and interpretation of data and critical revisions of the manuscript for important intellectual content. All authors approved the version to be published. KI is a guarantor of this work. Corresponding authors: KI.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We obtained approval from the ethics committee of Gifu University Graduate School of Medicine (Approval number 2020-201). The patient and her relatives provided written consent to participate in genetic screening for hereditary hypercalcemia.
Consent to publication
After she became at 18 years old, we obtained written informed consent of this case report from both herself and her parent. Genetic testing was carried out after counseling by a clinical genetic specialist. Her parents and her brothers were above 20 years old and we obtained written informed consent of this case report from themselves. We also obtained written informed consent for genetic test for her and her families. The patient and her families also gave written consent for their personal or clinical details along with any identifying images to be published in this study.
Competing interests
All authors have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sumida, A., Iizuka, K., Kato, T. et al. A case of familial hypocalciuric hypercalcemia type 1 due to CASR p.Pro55Leu mutation. BMC Endocr Disord 22, 164 (2022). https://doi.org/10.1186/s12902-022-01077-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-022-01077-5