
Abstract
Background
Recent research suggests that periodontitis can increase the risk of chronic obstructive pulmonary disease (COPD). In this study, we performed two-sample Mendelian randomization (MR) and investigated the causal effect of periodontitis (PD) on the genetic prediction of COPD. The study aimed to estimate how exposures affected outcomes.
Methods
Published data from the Gene-Lifestyle Interaction in the Dental Endpoints (GLIDE) Consortium’s genome-wide association studies (GWAS) for periodontitis (17,353 cases and 28,210 controls) and COPD (16,488 cases and 169,688 controls) from European ancestry were utilized. This study employed a two-sample MR analysis approach and applied several complementary methods, including weighted median, inverse variance weighted (IVW), and MR-Egger regression. Multivariable Mendelian randomization (MVMR) analysis was further conducted to mitigate the influence of smoking on COPD.
Results
We chose five single-nucleotide polymorphisms (SNPs) as instrumental variables for periodontitis. A strong genetically predicted causal link between periodontitis and COPD, that is, periodontitis as an independent risk factor for COPD was detected. PD (OR = 1.102951, 95% CI: 1.005–1.211, p = 0.039) MR-Egger regression and weighted median analysis results were coincident with those of the IVW method. According to the sensitivity analysis, horizontal pleiotropy’s effect on causal estimations seemed unlikely. However, reverse MR analysis revealed no significant genetic causal association between COPD and periodontitis. IVW (OR = 1.048 > 1, 95%CI: 0.973–1.128, p = 0.2082) MR Egger (OR = 0.826, 95%CI:0.658–1.037, p = 0.1104) and weighted median (OR = 1.043, 95%CI: 0.941–1.156, p = 0.4239). The results of multivariable Mendelian randomization (MVMR) analysis, after adjusting for the confounding effect of smoking, suggest a potential causal relationship between periodontitis and COPD (P = 0.035).
Conclusion
In this study, periodontitis was found to be independent of COPD and a significant risk factor, providing new insights into periodontitis-mediated mechanisms underlying COPD development.
Similar content being viewed by others


Introduction
Periodontitis (PD) is a frequent chronic inflammatory disorder with clinical manifestations of radiologically assessed alveolar bone loss, accompanying clinical attachment loss, gingival bleeding, and periodontal pocket formation [1]. Host immunophysiological disruption and genetic factors play an important role in the development of PD [2, 3]. Recently, genetic susceptibility factors were found to play a role in the development of PD. For instance, Matthias Munz et al. have identified a correlation between genetic variants in the loci SIGLEC5 and DEFA1A3 and the risk of PD [4].
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory disease characterized by restriction of expiratory airflow and persistent lung parenchymal obstruction, along with emphysematous lung destruction [5]. Changes in lung microecology, autoimmune components of the disease, and environmental risk factors together contribute to the development of COPD. In addition to the above, genetic susceptibility has been implicated in COPD [6].
Although COPD primarily affects the lungs, it is now considered as a multi-component disease, often co-existing with other disorders such as PD [7]. Similarities between the two diseases exist, with neutrophil infiltration, altered protease/anti-protease, and redox state balance playing a key role [8]. Several observational studies show an independent and significant link between PD and COPD. For example, some retrospective studies show that the development of PD increases the risk of death among COPD patients [9, 10]. Recently, a systematic review including two cross-sectional studies and a case-control study found that frequent COPD exacerbations were strongly associated with PD [11]. However, in contrast, Arianne K Baldomero and Zhou X, concluded that periodontal health status was not associated with a worsened COPD state [12, 13]. However, observational studies using cross-sectional or case-control methods suffer from the disadvantages of bias, confounding factors, and reverse causality. Therefore, clarifying the genetic causal relationship between PD and COPD risk is important for assessing the pathogenesis of COPD.
Mendelian randomization (MR) is a causal analysis method that exploits natural randomization in the generation of individual genetic constituents. MR uses genetic data including single nucleotide polymorphisms (SNPs) that are related to exposure, as instrumental variables to assess the causality of the association between exposure or risk factors and the outcome of interest. MR analysis can overcome the above-mentioned limitations of observational studies in causality investigations and can compensate for the disadvantages of randomized controlled trials (RCTs) including high human and material consumption and ethical challenges. A new perspective unlike traditional observational studies and RCTs is presented [14]. We conducted a two-sample MR study with data from genome-wide association studies (GWAS) to investigate if there is a genetically predicted causal relationship between PD and COPD, thus facilitating the prevention and treatment of these patients.
Methods
Study design
MR is a causal analysis method based on Mendel’s segregation and independent assortment laws [15]. This study uses a two-sample MR design to estimate the causal influence of exposure on the outcome using GWAS summary data from two independent studies. Three assumptions underpin the MR design. First, genetic variations are consistently linked to exposure. Second, no confounders are related to genetic variations. Third, apart from the exposure, genetic variations do not affect the outcomes. MR is a useful technique in genetic epidemiology because it provides a trustworthy approach to investigating causal inference.
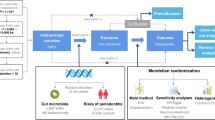
Our study design, with its 3 above-mentioned key components, is shown in Fig. 1.

The schematic depiction of two-sample Mendelian randomization design. Valid genetic instrumental variations that satisfy the three assumptions required for Mendelian randomization
Data sources
Data on the genetic association of SNPs with PD were obtained from the latest and largest GWAS meta-analysis of the Gene-Lifestyle Interaction in the Dental Endpoints (GLIDE) Consortium, with a total sample size of 17,353 clinical cases and 28,210 controls (PMID:31235808) [16]. Patients with PD were as defined based on the Centers for Disease and Control and Prevention/American Academy of Periodontology (CDC/AAP) [17] or Community Periodontal Index (CPI) or by diagnostic classification of PD reported by study participants [16]. COPD statistics were obtained from the publicly available GWAS dataset from European ancestry, including 16,488 cases and 169,688 controls (PMID:33106845) [18]. We conducted a two-sample MR study using PD and COPD as the exposure and outcome, respectively.
Selection of the instrumental variables
To satisfy the first assumption, we screened SNPs based on a P-value of 5 × 10−6 and used them as instrumental variables to identify independent SNPs associated substantially with PD. Second, we utilized the clumping approach with R2 < 0.001 and a window size of 10,000 kb to eliminate SNPs showing a strong linkage disequilibrium (LD). SNPs with effect allele frequency (EAF) of more than 0.01 were eliminated as well. The first condition was further verified by calculating the F-statistic and the explained phenotypic variance. IVs with an F-statistic of less than 10 were deemed weak instruments and eliminated from MR analysis [19]. To ensure that the second assumption was met, we used the PhenoScanner database for formerly reported correlations between instrumental SNPs (and LD proxies) and a potential confounding factor. These confounders included obesity and type 2 diabetes, BMI, alcohol, smoking, and rheumatoid arthritis. We used (P < 1e-5, r2 > 0.8) selection thresholds to strengthen instrument analyses [20]. Considering the third assumption to ensure that genetic variants did not influence the outcome through any variable other than exposure, the instrumental variables screened in PD were also selected in COPD. Finally, we reconciled the exposure and result datasets. Furthermore, a lack of instrument-exposure relationship may have diminished the validity of the MR relevance assumption.
Statistical analysis
To analyze the potential causal relationship between exposure and outcome, we used the R package, TwoSampleMR (0.5.6) [21]. In this study, to quantify the causal effects of exposure on its outcome, we used several complementary approaches, including the inverse variance weighted (IVW), MR-Egger regression [22], and weighted median [23]. IVW approach used to pool Wald ratios was the primary method of analysis [24]. IVW is a weighted regression of the combined SNP-exposure and SNP-outcome associations. If all of the IVs are valid or horizontal pleiotropy is balanced, its outcome is robust. Finally, given the strong correlation between smoking and COPD, we also conducted multivariable Mendelian randomization (MVMR) analysis to eliminate the interference of smoking-related phenotypes.
We used the Cochran’s Q-statistic from the IVW and MR Egger methods to test for heterogeneity (variability in causal estimates derived for each SNP) between the causal estimates of individual SNPs [25]. MR-Egger regression was used to assess the likelihood of horizontal pleiotropy. The intercept in the MR-Egger test represented the average direct impact of a variant on the results, while the slope represented the potential causal effect. To find horizontal pleiotropic outliers and identify whether there were considerable differences in the causal effects before and after outlier filtering, MR-PRESSO (pleiotropy residual sum and outlier) was employed [26]. To determine whether a specific SNP instrument was driving the discrepancy in computed residual sum of squares (RSS) versus simulated expectations, the “leave-one-out” technique was utilized.
Results
Instrumental variable selection for PD and COPD
Genetic variation is used as a proxy measure for risk factors in the MR approach to estimate the causal impact of the concerned risk factor. Fifty-nine independent SNPs associated with PD were identified in the latest GWAS dataset (p < 5 × 10−6). After the removal of LD (R2 < 0.001 and window size, 10,000 kb) and effect alleles (EAF > 0.01), eight SNPs remained. The details of these eight SNPs with respect to PD are shown in Supplementary Table S1. Considering the common confounders of PD, we generated associations of SNPs based on the PhenoScanner search, excluding a confounder BMI-related SNP (rs2921075). Subsequently, SNPs that were absent and/or significant in GWAS for COPD were excluded (rs138868497); six of these SNPs were included in the analysis. Moreover, due to the difficulties in identifying the effect alleles for the exposure and outcome, palindromic SNPs (i.e., an allele of an SNP consisting of a base and its complementary base) were removed (rs4811024). Ultimately, valid instrumental variables that met the three MR conditions stated above were selected. Five SNPs were included in the MR analysis, namely rs10143801, rs151226594, rs73155039, rs76734229, and rs9954920 (Table 1). Finally, we reversed the exposure and outcome, considering COPD as the exposure and periodontitis as the outcome. Using the same selection criteria, we identified 32 SNPs for analyzing the reverse causal relationship between COPD and periodontitis.
All F-statistic values were > 10, indicating no weak instrument bias.
Sensitivity analysis supporting the causal association between PD and COPD
We employed the MR-Egger approach for measuring heterogeneity among the causal assessments of individual SNPs. Cochran’s Q-statistic showed no substantial indication of heterogeneity between SNPs (P = 0.9204 > 0.05). Among the IVW estimates, no substantial heterogeneity variability between Wald ratios was found. Analysis of the MR-Egger intercept revealed no directional pleiotropy (Table 2). The funnel plot also supported the above findings (Fig. S1).
Furthermore, the leave-one-out analysis showed no significant differences in PD and COPD causal estimations, indicating that none of the discovered causative links were driven by a single IV. The causal links between PD and COPD are depicted in the leave-one-out graphic (Fig. 2).

MR analysis and leave-one-out analysis of the causal effect of PD on COPD susceptibility (A) A scatter plot depicting the causal relationships between PD and COPD using various MR methods. Each line’s slope corresponds to the expected MR effect for each method. B The causal links between periodontitis and COPD are depicted using a leave-one-out plot. The leave-one-out figure depicted how the removal of a single variant altered the causal estimations (point with horizontal line) for the effect of periodontitis on COPD
The causal associations of PD with COPD
We used IVW, weighted median, and MR-Egger approaches for MR analysis. The results of MR analysis favored a significant causal association of PD with COPD. The IVW (OR = 1.102951 > 1, 95%CI: 1.005–1.211, p = 0.039) method showed statistical significance for PD as a notable risk factor for COPD. MR Egger (OR = 1.163, 95%CI:1.021–1.324, p = 0.1071) and weighted median (OR = 1.122, 95%CI: 0.993–1.268, p = 0.0641) methods showed results consistent with those of IVW. Estimated causal effects between PD and COPD using different MR methods are presented in Table 3 and Figs. 2 and 3.

Comparison of Mendelian randomization results obtained using various approaches. PD: periodontitis; COPD: chronic obstructive pulmonary disease; IVW: inverse variance weighted; OR: odds ratio; CL: confidence interval
The causal associations of COPD with PD
We reversed the exposure and outcome, considering COPD as the exposure and periodontitis as the outcome. Using the same selection criteria, we identified 32 SNPs for analyzing the reverse causal relationship between COPD and periodontitis. The MR results indicate no significant genetically predicted association between COPD and periodontitis. IVW (OR = 1.048 > 1, 95%CI: 0.973–1.128, p = 0.2082) MR Egger (OR = 0.826, 95%CI:0.658–1.037, p = 0.1104) and weighted median (OR = 1.043, 95%CI: 0.941–1.156, p = 0.4239)(Table 4) According to Cochran’s Q-statistic, there was no notable evidence of heterogeneity among the SNPs. (MR-Egger p = 0.260 IVW p = 0.132) Additionally, the MR-Egger intercept test results effectively ruled out any directional pleiotropy. (p = 0.462) Furthermore, the leave-one-out sensitivity analysis demonstrated that no specific SNP significantly contributed to the association between COPD and PD.(Supp.Fig.S2-S4).
MVMR analysis
Considering the potential contribution of smoking to the progression of COPD, we employed multivariable Mendelian randomization (MVMR) to examine significant associations, adjusting for the impact of smoking on COPD. The results of the multivariable MR analysis indicate that after adjusting for the confounding effects of smoking, a genetic causal relationship between periodontitis and COPD may be inferred. (P = 0.035<0.05)(Table 5).
Discussion
This is the first report of a two-sample MR analysis for determining the genetic causal relationship between PD and COPD. The MR study provided convincing evidence that PD is genetically causally associated with COPD and is an independent risk factor for COPD development.
The first association between dental health and COPD in community-dwelling populations was discovered in a study of 23,808 people by the National Health and Nutrition Examination Survey I (NHANESI) [27]. Observational studies suggest that acute exacerbation of PD is a key risk factor for COPD progression and is associated with high mortality in COPD patients [28,29,30]. Frank A Scannapieco and coworkers found that the more severe the periodontal attachment loss, the greater the risk of COPD (odds ratio: 1.35; 95% confidence interval: 1.07–1.71) [31]. Hayes et al. also found that PD, as measured by alveolar bone loss assessed by periapical radiographs, was an independent risk factor for COPD [32]. Niamh Kelly and ZeSheng Wu et al. also suggested that poor periodontal health was associated with worsened COPD [11, 33]. Recently, Liu Shu qin et al. identified a potential genetic crosstalk between PD and COPD [34]. PD was further identified as an important and independent risk factor for COPD based on a meta-analysis of 14 observational studies [35]. Finally, A meta-analysis comprising 75 survey studies revealed a significant positive correlation between periodontitis and COPD [36]. These findings provide preliminary evidence that PD is an important factor that promotes COPD.
The results of this MR study strongly supported a genetically predicted causal relationship between PD and COPD and demonstrated that the former is an important risk factor for the latter. Therefore, the potential pathophysiological relationship between PD and COPD warrants further investigation.
We believe that the causal relationship between these two diseases may be closely related to the following reasons. First, oral flora is an important factor in the causal relationship between PD and COPD. Previous studies implicated oral bacteria in lung infections. Through various masticatory motions, dental plaque shed into saliva may change the respiratory epithelium, allowing pulmonary pathogen colonies to adhere strongly and grow [37]. Andreea C Didilescu et al. showed that dental plaque may serve as a reservoir for respiratory bacteria [38], crucial for the advancement of COPD. Studies have identified common pathogens between PD and COPD, including Porphyromonas gingivalis, Tannerella forsythia, Haemophilus, and Treponema denticola [39]. Among these, Porphyromonas gingivalis is closely linked to the development and progression of periodontal disease, being a gram-negative anaerobic bacterium commonly colonizing periodontal pockets [40]. Nan Feng et al. found that it could migrate to the lungs, alter the pulmonary microbiota, and exacerbate chronic obstructive pulmonary disease (COPD) [41]. Improved dental health may reasonably lower morbidity in COPD patients [42]. Second, both diseases are also associated with inflammatory mediators. Local inflammation in the periodontium results in the release of several proinflammatory cytokines into the bloodstream, including interleukins IL-6, IL-1α, and IL-1β, interferon IFN-γ, and tumor necrosis factor (TNF-α) [43]. These inflammatory factors may be connected to respiratory disease infections. In animal and cell studies, neutrophils have been linked to the onset and progression of COPD, by releasing inflammatory mediators like neutrophil elastase and matrix metalloproteinases (MMPs) [10]. According to a review by Hajishengallis, diseases characterized by aberrant neutrophil functions have a higher associated frequency of PD, including neutrophil deficiency and autoimmune neutropenia [38]. Epidemiological studies have found evidence to support this relationship. Finally, it is noteworthy that Kaixin Xiong et al. first discovered the crucial role of the γδ T-M2 immune mechanism in mediating periodontitis-promoted COPD [44]. Additionally, Shuqin Liu et al. identified EPB41L4A-AS1 as a potential cross-interacting gene between the two diseases. Its downregulation activates the nuclear factor kappa B (NF-κB) signaling pathway and enhances inflammatory responses, which also plays a pivotal role in the pathogenesis of periodontitis-promoted COPD [34].
Our MR analysis has numerous advantages. First, environmental factors and behavior do not affect genetic variation. As a result, MR analysis decreases residual confounders and other biases, effectively reducing reverse causality [14]. To evaluate the possible genetic causal link between PD and COPD, we used the most recent and largest GWAS dataset for PD and COPD from a European-descent population, therefore minimizing the influence of population stratification. Additionally, MR also has the advantages of being cost-effective and having few ethical concerns [45]. Lastly, we used a two-sample design to estimate the link between genetic variant exposure and genetic variant outcomes from two independent comparable populations, yielding greater statistical power [46].
Limitations
Some limitations also warrant consideration. First, the SNPs used were all from individuals of European ancestry. Such studies for other ethnicities are warranted. Currently, MR studies often assess the lifelong effects of risk factors on outcomes, and it is difficult to reveal the causal effects across the stages of disease development. Finally, given the varied definitions of PD employed in different studies, GWAS for consistent SNPs in PD is difficult.
Conclusion
In conclusion, this study supports a genetically predicted association between PD and COPD through MR Analysis and considers the former to be a risk factor for the latter. Although the cause underlying this relationship needs to be studied further, we have provided genetic evidence that PD is linked to COPD. In the future, this genetic tool is expected to be valuable for preventing and treating COPD. Maintaining good dental hygiene may contribute to reducing the risk of developing chronic obstructive pulmonary disease (COPD). However, further research is needed to confirm these observational findings and explore potential mechanisms.
Availability of data and materials
The periodontitis summary statistic data are available at https://data.bris.ac.uk/data/dataset/.
The COPD summary data are available at https://www.ebi.ac.uk/gwas/studies/GCST90016594
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- PD:
-
Periodontitis
- MR:
-
Mendelian randomization
- IVW:
-
Inverse variance weighted
- SNPs:
-
Single-nucleotide polymorphisms
References
Baumeister SE, Nolde M, Holtfreter B, Baurecht H, Gläser S, Kocher T, et al. Periodontitis and pulmonary function: a Mendelian randomization study. Clin Oral Investig. 2021;25(8):5109–12.
Zhang J, Lin S, Luo L, Zhang Q, Jiao Y, Liu W. Psychological stress: neuroimmune roles in periodontal disease. Odontology. 2023;111(3):554–64.
Offenbacher S, Divaris K, Barros SP, Moss KL, Marchesan JT, Morelli T, et al. Genome-wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum Mol Genet. 2016;25(10):2113–29.
Munz M, Willenborg C, Richter GM, Jockel-Schneider Y, Graetz C, Staufenbiel I, et al. A genome-wide association study identifies nucleotide variants at SIGLEC5 and DEFA1A3 as risk loci for periodontitis. Hum Mol Genet. 2017;26(13):2577–88.
Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–42.
Silverman EK. Genetics of COPD. Annu Rev Physiol. 2020;10(82):413–31.
Negewo NA, Gibson PG, McDonald VM. COPD and its comorbidities: impact, measurement and mechanisms. Respirol Carlton Vic. 2015;20(8):1160–71.
Pathak JL, Yan Y, Zhang Q, Wang L, Ge L. The role of oral microbiome in respiratory health and diseases. Respir Med. 2021;185:106475. https://doi.org/10.1016/j.rmed.2021.106475.
Qian Y, Yuan W, Mei N, Wu J, Xu Q, Lu H, et al. Periodontitis increases the risk of respiratory disease mortality in older patients. Exp Gerontol. 2020;133:110878.
Sczepanik FSC, Grossi ML, Casati M, et al. Periodontitis is an inflammatory disease of oxidative stress: we should treat it that way. Periodontol. 2020;84(1):45–68.
Kelly N, Winning L, Irwin C, Lundy FT, Linden D, McGarvey L, et al. Periodontal status and chronic obstructive pulmonary disease (COPD) exacerbations: a systematic review. BMC Oral Health. 2021;21:425.
Zhou X, Wang J, Liu W, Huang X, Song Y, Wang Z, et al. Periodontal status and microbiologic pathogens in patients with chronic obstructive pulmonary disease and periodontitis: a case-control study. Int J Chron Obstruct Pulmon Dis. 2020;15:2071–9.
Baldomero AK, Siddiqui M, Lo CY, Petersen A, Pragman AA, Connett JE, et al. The relationship between oral health and COPD exacerbations. Int J Chron Obstruct Pulmon Dis. 2019;14:881–92.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98.
Emdin CA, Khera AV, Kathiresan S. Mendelian Randomization. JAMA. 2017;318(19):1925–6.
Shungin D, Haworth S, Divaris K, Agler CS, Kamatani Y, Keun Lee M, et al. Genome-wide analysis of dental caries and periodontitis combining clinical and self-reported data. Nat Commun. 2019;10(1):2773.
Page RC, Eke PI. Case definitions for use in population-based surveillance of periodontitis. J Periodontol. 2007;78(7 Suppl):1387–99.
Kim W, Prokopenko D, Sakornsakolpat P, Hobbs BD, Lutz SM, Hokanson JE, et al. Genome-wide gene-by-smoking interaction study of chronic obstructive pulmonary disease. Am J Epidemiol. 2021;190(5):875–85.
Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64.
Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat Genet. 2020;52(7):740–7.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-base platform supports systematic causal inference across the human phenome. eLife. 2018;30(7):e34408.
Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181(4):251–60.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–906.
Bae SC, Lee YH. Causal association between periodontitis and risk of rheumatoid arthritis and systemic lupus erythematosus: a Mendelian randomization. Z Für Rheumatol. 2020;79(9):929–36.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Scannapieco FA, Papandonatos GD, Dunford RG. Associations between oral conditions and respiratory disease in a national sample survey population. Ann Periodontol. 1998;3(1):251–6.
Saito M, Shimazaki Y, Yoshii S, Takeyama H. Periodontitis and the incidence of chronic obstructive pulmonary disease: a longitudinal study of an adult Japanese cohort. J Clin Periodontol. 2023;50(6):717–26.
Sundh J, Tanash H, Arian R, et al. Advanced dental cleaning is associated with reduced risk of COPD exacerbations - a randomized controlled trial. Int J Chron Obstruct Pulmon Dis. 2021;16:3203–15. Published 2021 Nov 25
Flattet Y, Garin N, Serratrice J, Perrier A, Stirnemann J, Carballo S. Determining prognosis in acute exacerbation of COPD. Int J Chron Obstruct Pulmon Dis. 2017;31(12):467–75.
Scannapieco FA, Bush RB, Paju S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease. A Systematic Review Ann Periodontol. 2003;8(1):54–69.
Hayes C, Sparrow D, Cohen M, Vokonas PS, Garcia RI. The association between alveolar bone loss and pulmonary function: the VA dental longitudinal study. Ann Periodontol. 1998;3(1):257–61.
Wu Z, Xiao C, Chen F, Wang Y, Guo Z. Pulmonary disease and periodontal health: a meta-analysis. Sleep Breath Schlaf Atm. 2022;26(4):1857–68.
Liu S, Fu Y, Ziebolz D, Li S, Schmalz G, Li F. Transcriptomic analysis reveals pathophysiological relationship between chronic obstructive pulmonary disease (COPD) and periodontitis. BMC Med Genet. 2022;15(1):130.
Zeng XT, Tu ML, Liu DY, Zheng D, Zhang J, Leng W. Periodontal disease and risk of chronic obstructive pulmonary disease: a meta-analysis of observational studies. PLoS One. 2012;7(10):e46508.
Adam M. Does periodontitis affect respiratory health? Evid Based Dent. 2023;24(3):102–3.
Scannapieco FA, Cantos A. Oral inflammation and infection, and chronic medical diseases: implications for the elderly. Periodontol. 2016;72(1):153–75.
Hajishengallis G. Interconnection of periodontal disease and comorbidities: evidence, mechanisms, and implications. Periodontol. 2022;89(1):9–18.
Pinto EH, Longo PL, de Camargo CCB, Dal Corso S, Lanza FDC, Stelmach R, et al. Assessment of the quantity of microorganisms associated with bronchiectasis in saliva, sputum and nasal lavage after periodontal treatment: a study protocol of a randomised controlled trial. BMJ Open. 2016;6(4):e010564.
Shi T, Wang J, Dong J, Hu P, Guo Q. Periodontopathogens Porphyromonas gingivalis and Fusobacterium nucleatum and their roles in the progression of respiratory diseases. Pathogens. 2023;12(9):1110.
Feng N, Han X, Peng D, et al. P. Gingivalis alters lung microbiota and aggravates disease severity of COPD rats by up-regulating Hsp90α/MLKL. J Oral Microbiol. 2024;16(1):2334588.
Apessos I, Voulgaris A, Agrafiotis M, Andreadis D, Steiropoulos P. Effect of periodontal therapy on COPD outcomes: a systematic review. BMC Pulm Med. 2021;21(1):92.
Liu J, Ouyang Y, Zhang Z, et al. The role of Th17 cells: explanation of relationship between periodontitis and COPD? Inflamm Res. 2022;71(9):1011–24.
Xiong K, Ao K, Wei W, et al. Periodontitis aggravates COPD through the activation of γδ T cell and M2 macrophage. mSystems. 2024;9(2):e0057223.
Song CJ, Bian MY, Lei LH, Chen LL. Mendelian randomization and its application in periodontitis. Zhonghua Kou Qiang Yi Xue Za Zhi Zhonghua Kouqiang Yixue Zazhi Chin J Stomatol. 2022;57(10):1072–8.
Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–52.
Acknowledgements
The authors acknowledge and thank the investigators of the original GWAS studies for sharing summary data used in this study.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 82001075), the Natural Science Foundation of the Science and Technology Department of Shanxi Province (Grant No. 202103021224232), and the Science and Technology Innovation Project of Higher Education Institutions of Shanxi Province (Grant No. 2022 L167).
Author information
Authors and Affiliations
Contributions
Bao-Ling Zhao, Xue-Jun Ge and Fei-Yan Yu conceived the study; Zhen-Ni Zhao, Rong Zhao and Qian-Qian Wang contributed to data collection and analysis; Bao-Ling Zhao and Fei-Yan Yu wrote the draft of the paper. Xue-Jun Ge revised and refined the manuscript; Bao-Ling Zhao, Fei-Yan Yu, Jia-Qi Yang, Yu-Kai Hao and Zi-Qian Zhang contribute to data interpretation; All authors approved the final draft and accepted the decision to submit the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval and consent were not specifically for this study as we used summary data that is publicly available.
Consent for publication
Not applicable.
Competing interests
All authors have declared that no conflicts of interest exist.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, BL., Yu, FY., Zhao, ZN. et al. Periodontal disease increases the severity of chronic obstructive pulmonary disease: a Mendelian randomization study. BMC Pulm Med 24, 220 (2024). https://doi.org/10.1186/s12890-024-03025-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-024-03025-6