
Abstract
Background
BCR::ABL1-like or Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL) was first reported in 2009. Ph-like ALL is characterized by gene signature similar to Philadelphia chromosome ALL, but without BCR::ABL1 fusions. Molecularly, Ph-like ALL is divided into seven categories, with CRLF2 and ABL-class rearrangements being the two most common subtypes, exhibiting alterations in distinct downstream signaling cascades.
Case presentation
We report a rare case of pediatric Ph-like ALL with concomitant CRLF2 and ABL1 rearrangements. CRLF2 was fused with P2RY8, its most common fusion partner, whereas ABL1 was fused with MYO18B, a novel fusion partner that has not been previously reported. The 4-year-old female patient was treated using the national multicenter CCCG-ALL-2020 protocol with the addition of dasatinib at the end of induction when ABL1 rearrangement was confirmed by RNA-seq. Morphologically and molecularly, the patient remained in continuous remission until the last follow-up. To the best of our knowledge, this is the first case of Ph-like ALL harboring two distinct rearrangement categories.
Conclusions
Our results identified that ABL1 rearrangement and CRLF2 rearrangement can coexist. The application of FISH, whole transcription sequencing, PCR can help us to have a more comprehensive understanding of ALL cytogenetics and molecular biology. Further studies are needed to explore the role of targeted therapies in such rare clinical scenarios.
Similar content being viewed by others
Background
BCR::ABL1-like or Philadelphia chromosome-like (Ph-like) acute lymphoblastic leukemia (ALL) is characterized by a poor early treatment response, high recurrence rate, and unfavorable clinical outcomes. The 5-year EFS/DFS of children Ph-like ALL varied widely across regions, ranging from around 30–90%. The 5-year EFS of adults Ph-like ALL were about 24% [1]. Ph-like ALL cases most commonly present with CRLF2 or ABL class rearrangements [2, 3]; however, Ph-like ALL with concomitant rearrangements of both CRLF2 and ABL1 has not been identified previously. Here, we report a rare pediatric case of Ph-like ALL with concomitant CRLF2 and ABL1 rearrangements. CRLF2 was fused with P2RY8, the most common fusion partner, whereas ABL1 was fused with MYO18B, a novel fusion partner that has not been previously reported. Studies have shown that ABL-class-rearranged ALL has a good therapeutic response to the tyrosine kinase inhibitors (TKI) imatinib or dasatinib [4,5,6,7,8], similar to Ph positive ALL. However, effective therapies for CRLF2 rearrangements are still under investigation. With the combination of chemotherapy and dasatinib, the patient has remained measurable residual disease (MRD)-negative for 18 months since therapy initiation.
Case presentation
A 4-year-old female was admitted to our hospital with intermittent fever and bilateral preauricular masses that persisted for 16 days. Approximately 16 days before admission, the painless preauricular lumps had grown steadily. Concomitantly, the patient presented with intermittent fever, exhibiting a maximum temperature of 38.8 °C. Her temperature normalized after intravenous penicillin administration at a local hospital. Before admission, the patient had no symptoms such as weariness, epistaxis, skin bleeding, or ostealgia. However, three kilograms of body weight were lost in the two weeks prior to admission. The patient had no history of illness. The patient had no specific personal and family history of similar illnesses. On admission, she was in a relatively stable condition but appeared pale. The size of the left preauricular was 2 by 2 cm and right preauricular was 2.5 by 2.5 cm, exhibiting moderate texture and was not tender. Abdominal palpation revealed hepatomegaly and splenomegaly 5 and 7 cm below the ribs, respectively. Physical examinations of the heart, lungs, and nervous system revealed negative results. White blood cell count was 5.4 × 109/L, lymphocytes were 51%, neutrophils were 19%, monocytes were 1%, blast cells were 25%, hemoglobin was 63 g/L, platelets were 36 × 109/L, reticulocytes were 0.14%, and absolute reticulocyte count was 0.0045 × 109/L on the complete blood count. Blood biochemical testing revealed normal renal function; alanine aminotransferase 42 U/L; aspartate aminotransferase 18 U/L; lactate dehydrogenase 157 U/L; and normal blood electrolyte levels. The results from the disseminated intravascular coagulation were normal. DNA tests for Epstein-Barr virus and Cytomegalovirus were negative. Abdominal ultrasonography revealed an enlarged liver and spleen. ALL was suspected based on a bone marrow smear, and the French-American-British classification was L2, with 88.5% lymphoblasts.
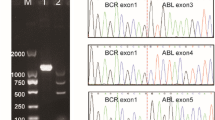
Pre-B ALL was identified by bone marrow flow cytometry based on the expression of CD19, cCD79a, CD22, CD10, TDT, CD34, HLA-DR, CD9, CD66c, and CD38, and lack of expression of CD3 or MPO. A positive MYO18B::ABL1 fusion gene (Fig. 1A), positive P2RY8::CRLF2 fusion gene (Fig. 1B), positive PTPN11 mutation (E76G, mutation frequency 31.2%), and positive ETV6 mutation (R105*, mutation frequency 1.6%) were identified using whole-transcriptome sequencing. The MYO18B::ABL1 fusion was verified by polymerase chain reaction (PCR) and sanger sequencing (Fig. 2). With positive rates of 49% and 85%, respectively, the ABL1 and CRLF2 probes demonstrated signal separation using fluorescence in situ hybridization (FISH) (Fig. 3). The probes used to detect the ABL1 break site were located at 9q34.12 and the CRLF2 break site were located at Xp22.33. CRLF2 overexpression was also observed, with an expression percentage of 99.4%. 47, XX, + 21[2]/47, idem,?t(1;9)(p34;q34),del [22](q11)[6]/46, XX[12] were the chromosomal karyotypes found. Analysis of karyotype results can sometimes be inaccurate and subjective, and our karyotype results suggest abnormalities of 9q.34 and 22q. ABL1 is located at 9q.34 and MYO18B is located at 22q. No IKZF1 gene deletions were found using multiplex ligation-dependent probe amplification. The methods and materials used for this patient’s assay are provided in the Supplementary Materials.

A positive MYO18B-ABL1 fusion gene and a positive P2RY8-CRLF2 fusion gene were suggested by whole-transcriptome sequencing. Diagram of the MYO18B/ABL1 fusion gene. The fracture point of MYO18B is located at chr22:26251330, and the fracture point of ABL1 is located on chr9:133738150, respectively. Diagram of the P2RY8-CRLF2 fusion gene. The fracture point of P2RY8 is located on chrX:1,655,814, and the fracture point of CRLF2 is located on chrX:1,321,529, respectively

Sanger sequencing and PCR electrophoresis of MYO18B/ABL1. (A) Sanger sequencing peak figure of MYO18B/ABL1. The fusion gene sequence is GCAGCGGGAGGCAGAGGCCAGCCGGCGGTGCATGGAGCT TCTACGTCTCCTCCGAGAGCCGCTTCAACACCCTGGCCGAG [GC]. (B) PCR electrophoresis shows that lane 3 is the PCR product of MYO18B-ABL1 while lane 4 is the negative control

FISH detection indicates signal separation in both ABL1 and CRLF2 (indicated by the white arrow). The ABL1 separation probe showed signal separation with a positive rate of approximately 49%, while the CRLF2 separation probe showed an abnormal signal mode with a positive rate of approximately 85%
Combined with the typical clinical signs and symptoms of the patient, the laboratory examination, especially the bone marrow morphology and genetic examination, confirmed the diagnosis of ALL beyond doubt. Based on the FISH and whole-transcriptome test results, we concluded that the child was Ph-like ALL.
Acute Lymphoblastic Leukemia-2020, an ongoing project of the China Children’s Cancer Group, was used to treat the patient once ALL was confirmed (CCCG-ALL-2020; www.chictr.org.cn; Date of registration: August 5, 2020; Registration number: ChiCTR2000035264). On day 6 after chemotherapy, the enlarged lymph nodes were no longer palpable, and on day 18, the enlarged liver was restored to its normal size. Following chemotherapy, routine complete blood count revealed that the red blood cells, white blood cells, and platelets in the peripheral blood were all stabilized around day 28. A satisfactory response to early chemotherapy was indicated on day 19 following chemotherapy, when the bone marrow smear showed complete remission and the flow cytometry (FCM)-MRD was less than 0.01%. On day 46, RT-PCR showed negative results for the MYO18B::ABL1 and P2RY8::CRLF2 fusion genes, and Sanger sequencing showed negative results for PTPN11 and ETV6 mutations. On day 28 of induction therapy, targeted therapy with dasatinib (80 mg/m2/day) was initiated. The patient developed severe serous cavity effusions and shortness of breath approximately 10 days after oral delivery. We immediately discontinued dasatinib, and these symptoms gradually improved. Fourteen days after discontinuation of dasatinib, a reduced dose of dasatinib (60 mg/m2/day) was re-administered at the beginning of consolidation therapy and continued until the end of consolidation therapy. At the beginning of continuous treatment, approximately four months after the initial treatment, we attempted to adjust the dose of dasatinib to 80 mg/m2/day, and the patient had no further adverse reactions.
It has been 18 months since the initiation of chemotherapy. Every four months, bone marrow smears, FCM-MRD, fusion gene qualification, and gene mutation quantification were performed, with consistently negative results (Fig. 4).

The treatment timeline of the patient. On day 28 of the induction therapy, targeted therapy of dasatinib (80 mg/m2/day) was added. The patient had severe serous cavity effusions and shortness of breath approximately 10 days after oral delivery. We immediately stopped the dasatinib administration and these symptoms gradually abated. Fourteen days after dasatinib was discontinued, a reduced dose of dasatinib (60 mg/m2/day) was added again at the beginning of the consolidation therapy and continued until the end of the consolidation therapy. Complete response at the genetic level of the bone marrow was achieved from 19 days after chemotherapy. DAS: dasatinib
Discussion and conclusions
BCR::ABL1-like or Ph-like ALL was first reported in 2009 [9, 10]. Ph-like and Ph + ALL have similar gene expression profiles and are both likely to be complicated by gene mutations in IKZF1, transcription factor 3 (E2A), early B-cell factor, and paired box 5 [5, 11, 12]. Mutations in PTPN11 and ETV6 were also detected in our patient. According to the literature, both PTPN11 and ETV6 mutations are common genetic lesions in Ph-like ALL [13,14,15]. Similar to BCL::ABL1-positive ALL, the prognosis of Ph-like ALL is worse than that of other types of B-cell precursor-ALL. According to the WHO classification of lymphopoietic tumors updated in 2022, Ph-like ALL is classified as a recurrent genetic abnormality of B-cell lymphoblastic leukemia/lymphoma [16] and is characterized by a poor early treatment response, high recurrence rate, and unfavorable clinical outcome.
The incidence of Ph-like ALL gradually increases with age and is approximately 10–15% in children, 20% in adolescents, and 20–27% in adults [5, 17, 18]. Ph-like ALL is more common in male patients. The male-to-female ratio is approximately 1.5:1 in children and 4:1 in adults [5]. In the CCCG-ALL-2020 protocol, methods used to screen for Ph-like ALL include PCR, FISH, and RNA-seq. These three methods are routine tests for all patients with ALL, according to our protocol.
Poor prognosis of patients with Ph-like ALL is associated with genetic abnormalities at the molecular level. The molecular biological abnormalities of Ph-like ALL mainly involve kinase and cytokine receptor activation. The kinase genes and cytokine receptors involved can be divided into the following major categories: CRLF2 rearrangement, ABL-class rearrangement (ABL1, ABL2, CSF1R, PDGFRA, and PDGFRB), JAK2 rearrangement, EPOR rearrangement, JAK/STAT aberrations (TSLP, IL2RB, and TYK2), RAS pathway mutations (KRAS, NRAS, NF1, PTPN11, CBL1, and BRAF), and other types of genetic abnormalities (NTRK3, PTK2B, BLNK, FLT3, FGFR1, DGKH) [2, 19, 20]. In addition, it has been reported that patients with positive ETV6::RUNX1, TCF3::PBX1, KMT2A-r, and BCR::ABL1 rearrangements usually do not have the above gene changes of Ph-like ALL simultaneously [21, 22].
Among the above categories, CRLF2 rearrangement is the most common genetic abnormality in Ph-like ALL at all ages, accounting for approximately 40–60% of cases [5]. The CRLF2 gene is located at Xp22.3 (OMIM: *300357)/Yp11.3 (OMIM: *400023), encoding the thymic stromal lymphopoietin receptor, which binds with the ligand of the α-chain heterodimer IL-7R to mediate the downstream signal transduction of lymphogenesis, anaphylaxis, and inflammation [23]. The vast majority of CRLF2 rearrangements have positive IGH::CRLF2 and P2RY8::CRLF2 fusion genes, and a few have CRLF2 point mutations [24,25,26,27]. Approximately 50–60% of patients with CRLF2 rearrangements are complicated by genetic abnormalities of the JAK or RAS pathways, such as NRAS, KRAS, PTPN11, and NF1, and often have loss of heterozygosity (LOH) of IKZF1 and iAMP21. CRLF2 rearrangements are also found in non-Ph-like ALL, specifically in Down Syndrome-associated ALL [28]. CRLF2 overexpression is associated with high MRD during early treatment and poor outcomes in high-risk ALL [29,30,31]. We carefully considered whether the JAK inhibitor ruxolitinib should be administered to our patient. Overall, ruxolitinib efficacy in patients with CRLF2-rearranged ALL remains unclear. Clinical studies on the ruxolitinib in patients with CRLF2 rearrangement ALL are limited, with only a small number of ongoing clinical trials in children and adults with Ph-like ALL (NCT02723994, NCT03571321, NCT03117751, NCT02420717, and NCT02723994) and a recently initiated European trial (EudraCT number 2020-005017-41). However, the final findings have not yet been published. Ruxolitinib is primarily used as an adjuvant treatment for ALL with abnormalities in JAK2 or the JAK-STAT pathways. RNA-seq clearly showed that there were no genetic aberrations in any genes in the JAK-STAT signaling pathway in our patient, and we presumed that the off-label use of ruxolitinib would not provide additional benefits. In addition, it has been suggested that P2RY8::CRLF2 could represent a secondary event because this fusion can be found concomitantly with other subtype-defining alterations such as iAMP21 or hyperdiploid ALL. In patients with ABL rearrangement, perhaps ABL1 fusion is more likely to be the key driver also [32,33,34].
The downstream signaling pathways JAK/STAT and P13K/AKT/MTORC1 were abnormally activated in CRLF2-rearranged cell lines. In IGH::CRLF2-positive cell lines, activation of the MTORC1 and RAS pathways plays a crucial role in their survival, but the survival of CRLF2-rearranged cell lines does not depend on JAK activation [35]. The MEK inhibitor trametinib and Akt inhibitor MK2206 have been shown to enhance the sensitivity of CRLF2-rearranged cell lines to hormones [36]. Moreover, the combined use of trametinib and gilteritinib can simultaneously inhibit MEK (RAS pathway) and receptor tyrosine kinases (RTKs) (P13K/AKT/MTORC pathway), playing an inhibitory role in CRLF2-rearranged cell lines [37].
ABL class rearrangement is also a common genetic abnormality in Ph-like ALL, accounting for approximately 10% of all Ph-like ALL cases. Children with Ph-like ALL have a higher proportion of ABL class rearrangements than adults [5]. Studies have shown that ABL class-rearranged ALL has a good therapeutic response to imatinib or dasatinib [4,5,6,7,8], similar to the response in patients with Ph + ALL. At present, many clinical trials have evaluated the efficacy and safety of dasatinib (ClinicalTrial.gov: NCT 02420717, NCT02883049, NCT03564470, and NCT02143414). Using the CCCG-ALL-2015 regimen, our randomized controlled study confirmed that dasatinib was significantly superior to imatinib in reducing recurrence in the bone marrow and central nervous system without increasing adverse reactions [8]. Therefore, in the CCCG-ALL-2020 regimen, dasatinib is recommended as the preferred drug for the targeted therapy of ABL-class Ph-like ALL.
A total of 13 ABL1 partner genes have been reported to date; however, the partner gene MYO18B (myosin-XVIIIb) identified in this case has not been previously reported [1, 3, 19]. MYO18B (OMIM, *607295) is located at 22q12.1. LOH of 22q and is common in patients with lung, ovarian, and colorectal cancers, suggesting that a tumor suppressor gene may be in this region. The research results of Nishioka et al. showed that the deletion, mutation, promoter methylation, and histone deacetylation of MYO18B in 22q lead to the reduction or inactivation of MYO18B activity, which is closely related to the occurrence of lung, ovarian, and colorectal cancers [38,39,40,41,42]. The low expression of MYO18B has been reported to be related to the poor prognosis of squamous cell carcinoma of the skin [43]. Therefore, MYO18B is currently considered a tumor suppressor gene.
Ph-like ALL with a positive MYO18B::ABL1 fusion gene has not yet been reported. To our knowledge, this is the first report of an MYO18B::ABL1 fusion gene in ALL. Because of the fusion of MYO18B and ABL1, the activity of MYO18B may be reduced. In this case, the 3’ breakpoint of ABL1 was at chr9:133738150 (GRCh38), which is a common breakpoint of ABL1 and can affect the function of ABL1. Therefore, we speculate that the formation of the MYO18B::ABL1 fusion gene may have been related to the occurrence of leukemia in this case. In addition, other genetic abnormalities relevant to the pathogenesis of leukemia were detected in our patient, including P2RY8::CRLF2 fusion gene-positivity, and mutations in PTPN11 and ETV6. We believe that these genetic abnormalities are potential therapeutic targets for our patient.
CRLF2 and ABL-class rearrangements are the two most common Ph-like ALL subtypes. To the best of our knowledge, patients with ALL featuring both CRLF2 and ABL1 rearrangements are rare. An adult patient with chronic myelogenous leukemia was reported to be BCR::ABL1-positive at initial diagnosis and presented with a new CRLF2 rearrangement during the B-lymphoblast phase [44]. Both FISH and RNA-seq in this case confirmed simultaneous CRLF2 and ABL1 rearrangements.
For patients with both CRLF2 and ABL-class rearrangements, it is also necessary for clinicians to consider the selection of reasonable targeted therapeutic drugs. In our report, our patient was treated with dasatinib in combination with chemotherapy, which mainly resulted for the following reasons. First, the targeting effect of dasatinib in ABL-class Ph-like ALL has been confirmed in several studies [1, 45, 46]. In addition, no clinically effective CRLF2-targeted drug is currently available. Third, after the induction therapy, flow cytometry of the bone marrow on day 19 showed that the FCM-MRD of the child was < 0.01%, suggesting a good early therapeutic response. Therefore, we selected dasatinib, which has a proven efficacy against ABL-class ALL, for targeted therapy. Moreover, guided by FCM-MRD and fusion-gene PCR monitoring, post-chemotherapy follow-up and bone marrow monitoring were performed. At present, 13 months after chemotherapy, the bone marrow smear, FCM-MRD, MYO18B::ABL1, and P2RY8::CRLF2 genes of the patient remain negative. We plan to regularly monitor and follow up on the bone marrow characteristics of the patient.
Whether children with Ph-like ALL require hemopoietic stem cell transplantation (HSCT) is a matter of concern for clinicians. At present, it is believed that chemotherapy combined with TKI can be considered for patients with continuously negative FCM-MRD results after induction therapy; however, the bone marrow smear, FCM-MRD, and genetic examination of children should be closely monitored. However, for children with Ph-like ALL whose FCM-MRD is not negative and high-risk patients with a 5-year disease-free survival of < 30%, HSCT may be needed [47]. Currently, the long-term relapse rate in patients with negative bone marrow FCM-MRD results prior to HSCT is relatively low. Therefore, it is recommended that combining chemotherapy with TKIs or administration of CAR-T cells to achieve bone marrow FCM-MRD negativity is recommended before HSCT [48]. After HSCT, it is also necessary to continuously monitor MRD and continue the oral administration of TKIs. However, the optimal course of TKI treatment after transplantation remains unclear. Some studies suggest that TKI withdrawal may be considered in patients with persistently negative MRD for 6 months to one year after HSCT [49, 50].
Overall, the incidence of Ph-like ALL in children is low; however, the prognosis for this specific malignancy is poor. Due to the extensive application in recent years of next-generation sequencing in clinical practice, more cases of Ph-like ALL have been reported. At present, only the ABL-class has TKIs as approved treatments. In contrast, targeted therapies for CRLF2 rearrangements, and other types of Ph-like ALL need to be evaluated in additional trials. More research and exploration are needed on the indications for HSCT in children with Ph-like ALL and whether they would benefit from HSCT.
Data availability
Sequence data that support the findings of this study have been deposited in the Sequence Read Archive with the primary accession code PRJNA1000109.
Abbreviations
- BCR:
-
Breakpoint cluster region
- MYO18B:
-
Myosin-XVIIIb
- CRLF2:
-
Cytokine receptor-like factor 2
- P2RY8:
-
Pyrimidinergic receptor p2y, g protein-coupled,8
- IGH:
-
Immunoglobulin heavy chain
- PTPN11:
-
Protein-tyrosine phosphatase, nonreceptor-type, 11
- ETV6:
-
ETS variant transcription factor 6
- IKZF1:
-
Ikaros family zinc finger 1
- ALL:
-
Acute lymphoblastic leukemia
- FISH:
-
Fluorescence in situ hybridization
- FCM:
-
Flow cytometry
- PCR:
-
Polymerase chain reaction
- MRD:
-
Measurable residual disease
- LOH:
-
Loss of heterozygosity
- CCCG-ALL-2020:
-
China Children’s Cancer Group Acute Lymphoblastic Leukemia 2020
- HSCT:
-
Hemopoietic stem cell transplantation
- TKI:
-
Tyrosine kinase inhibitors
- CAR-T:
-
Chimeric antigen receptor T cell
- DAS:
-
Dasatinib
References
Yadav V, Ganesan P, Veeramani R, Kumar VD. Philadelphia-Like Acute Lymphoblastic Leukemia: a systematic review. Clin Lymphoma Myeloma Leuk. 2021;21(1):e57–65.
Jain S, Abraham A. BCR-ABL1-like B-Acute Lymphoblastic Leukemia/Lymphoma: a Comprehensive Review. Arch Pathol Lab Med. 2020;144(2):150–5.
Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood. 2017;130(19):2064–72.
Maese L, Tasian SK, Raetz EA. How is the Ph-like signature being incorporated into ALL therapy? Best Pract Res Clin Haematol. 2017;30(3):222–8.
Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15.
Tanasi I, Ba I, Sirvent N, Braun T, Cuccuini W, Ballerini P, et al. Efficacy of tyrosine kinase inhibitors in Ph-like acute lymphoblastic leukemia harboring ABL-class rearrangements. Blood. 2019;134(16):1351–5.
Roberts KG, Yang YL, Payne-Turner D, Lin W, Files JK, Dickerson K, et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017;1(20):1657–71.
Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin Cancer Biol. 2022;84:144–52.
Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10(2):125–34.
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–80.
Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–4.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Martínez-Anaya D, Moreno-Lorenzana D, Reyes-León A, Juárez-Figueroa U, Dean M, Aguilar-Hernández MM et al. Characterization of Philadelphia-like Pre-B Acute Lymphoblastic Leukemia: experiences in Mexican Pediatric patients. Int J Mol Sci. 2022;23(17).
Iacobucci I, Roberts KG. Genetic alterations and therapeutic targeting of Philadelphia-Like Acute Lymphoblastic Leukemia. Genes (Basel). 2021;12(5).
Shiraz P, Payne KJ, Muffly L. The current genomic and molecular Landscape of Philadelphia-like Acute Lymphoblastic Leukemia. Int J Mol Sci. 2020;21(6).
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: lymphoid neoplasms. Leukemia. 2022;36(7):1720–48.
Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, et al. High frequency and poor outcome of Philadelphia chromosome-like Acute lymphoblastic leukemia in adults. J Clin Oncol. 2017;35(4):394–401.
Roberts KG, Pei D, Campana D, Payne-Turner D, Li Y, Cheng C, et al. Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J Clin Oncol. 2014;32(27):3012–20.
Pui CH, Roberts KG, Yang JJ, Mullighan CG. Philadelphia chromosome-like Acute Lymphoblastic Leukemia. Clin Lymphoma Myeloma Leuk. 2017;17(8):464–70.
Plotka A, Lewandowski K. BCR/ABL1-Like Acute Lymphoblastic Leukemia: from Diagnostic approaches to molecularly targeted therapy. Acta Haematol. 2022;145(2):122–31.
Boer JM, Marchante JR, Evans WE, Horstmann MA, Escherich G, Pieters R, et al. BCR-ABL1-like cases in pediatric acute lymphoblastic leukemia: a comparison between DCOG/Erasmus MC and COG/St. Jude signatures. Haematologica. 2015;100(9):e354–7.
Yoda A, Yoda Y, Chiaretti S, Bar-Natan M, Mani K, Rodig SJ, et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010;107(1):252–7.
Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012;120(4):833–42.
Cario G, Leoni V, Conter V, Baruchel A, Schrappe M, Biondi A. BCR-ABL1-like acute lymphoblastic leukemia in childhood and targeted therapy. Haematologica. 2020;105(9):2200–4.
Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009;41(11):1243–6.
Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–98.
Roberts KG, Mullighan CG. Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat Rev Clin Oncol. 2015;12(6):344–57.
Li Z, Chang TC, Junco JJ, Devidas M, Li Y, Yang W, et al. Genomic landscape of Down syndrome-associated acute lymphoblastic leukemia. Blood. 2023;142(2):172–84.
Wang Y, Li J, Xue TL, Tian S, Yue ZX, Liu SG, et al. Clinical, biological, and outcome features of P2RY8-CRLF2 and CRLF2 over-expression in pediatric B-cell precursor acute lymphoblastic leukemia according to the CCLG-ALL 2008 and 2018 protocol. Eur J Haematol. 2023;110(6):669–79.
Moorman AV, Schwab C, Ensor HM, Russell LJ, Morrison H, Jones L, et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30(25):3100–8.
Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a children’s Oncology Group study. Blood. 2012;119(15):3512–22.
Vesely C, Frech C, Eckert C, Cario G, Mecklenbräuker A, Zur Stadt U, et al. Genomic and transcriptional landscape of P2RY8-CRLF2-positive childhood acute lymphoblastic leukemia. Leukemia. 2017;31(7):1491–501.
Morak M, Attarbaschi A, Fischer S, Nassimbeni C, Grausenburger R, Bastelberger S, et al. Small sizes and indolent evolutionary dynamics challenge the potential role of P2RY8-CRLF2-harboring clones as main relapse-driving force in childhood ALL. Blood. 2012;120(26):5134–42.
Tsai AG, Yoda A, Weinstock DM, Lieber MR. T(X;14)(p22;q32)/t(Y;14)(p11;q32) CRLF2-IGH translocations from human B-lineage ALLs involve CpG-type breaks at CRLF2, but CRLF2/P2RY8 intrachromosomal deletions do not. Blood. 2010;116(11):1993–4.
Zhang Q, Shi C, Han L, Jain N, Roberts KG, Ma H, et al. Inhibition of mTORC1/C2 signaling improves anti-leukemia efficacy of JAK/STAT blockade in CRLF2 rearranged and/or JAK driven Philadelphia chromosome-like acute B-cell lymphoblastic leukemia. Oncotarget. 2018;9(8):8027–41.
Meyer LK, Delgado-Martin C, Maude SL, Shannon KM, Teachey DT, Hermiston ML. CRLF2 rearrangement in Ph-like acute lymphoblastic leukemia predicts relative glucocorticoid resistance that is overcome with MEK or akt inhibition. PLoS ONE. 2019;14(7):e0220026.
Sasaki K, Yamauchi T, Semba Y, Nogami J, Imanaga H, Terasaki T, et al. Genome-wide CRISPR-Cas9 screen identifies rationally designed combination therapies for CRLF2-rearranged Ph-like ALL. Blood. 2022;139(5):748–60.
Nishioka M, Kohno T, Tani M, Yanaihara N, Tomizawa Y, Otsuka A, et al. MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc Natl Acad Sci U S A. 2002;99(19):12269–74.
Yanaihara N, Nishioka M, Kohno T, Otsuka A, Okamoto A, Ochiai K, et al. Reduced expression of MYO18B, a candidate tumor-suppressor gene on chromosome arm 22q, in ovarian cancer. Int J Cancer. 2004;112(1):150–4.
Tani M, Ito J, Nishioka M, Kohno T, Tachibana K, Shiraishi M, et al. Correlation between histone acetylation and expression of the MYO18B gene in human lung cancer cells. Genes Chromosomes Cancer. 2004;40(2):146–51.
Yokota J, Kohno T. Molecular footprints of human lung cancer progression. Cancer Sci. 2004;95(3):197–204.
Nakano T, Tani M, Nishioka M, Kohno T, Otsuka A, Ohwada S, et al. Genetic and epigenetic alterations of the candidate tumor-suppressor gene MYO18B, on chromosome arm 22q, in colorectal cancer. Genes Chromosomes Cancer. 2005;43(2):162–71.
Cao C, Zhang C, Sun Y, Mu Z, Shen Q. Myosin18B predicts favorable prognosis of cutaneous squamous-cell carcinoma. Genes Genomics. 2021;43(4):371–8.
Thakral B, Jain N, Tang G, Konoplev S, Vega F, Medeiros LJ, et al. From the archives of MD Anderson Cancer Center: concurrent BCR-ABL1 and CRLF2 rearrangements in B-lymphoblast phase of chronic myeloid leukemia. Ann Diagn Pathol. 2021;53:151767.
Inaba H, Azzato EM, Mullighan CG. Integration of Next-Generation sequencing to treat Acute Lymphoblastic Leukemia with Targetable lesions: the St. Jude Children’s Research Hospital Approach. Front Pediatr. 2017;5:258.
Cario G, Leoni V, Conter V, Attarbaschi A, Zaliova M, Sramkova L, et al. Relapses and treatment-related events contributed equally to poor prognosis in children with ABL-class fusion positive B-cell acute lymphoblastic leukemia treated according to AIEOP-BFM protocols. Haematologica. 2020;105(7):1887–94.
Tasian SK, Peters C. Targeted therapy or transplantation for paediatric ABL-class Ph-like acute lymphocytic leukaemia? Lancet Haematol. 2020;7(12):e858–9.
Vettenranta K, Dobšinská V, Kertész G, Svec P, Buechner J, Schultz KR. What is the role of HSCT in Philadelphia-Chromosome-Positive and Philadelphia-chromosome-like ALL in the tyrosine kinase inhibitor era? Front Pediatr. 2021;9:807002.
Warraich Z, Tenneti P, Thai T, Hubben A, Amin H, McBride A, et al. Relapse Prevention with tyrosine kinase inhibitors after allogeneic transplantation for Philadelphia chromosome-positive Acute Lymphoblast Leukemia: a systematic review. Biol Blood Marrow Transpl. 2020;26(3):e55–64.
Giebel S, Czyz A, Ottmann O, Baron F, Brissot E, Ciceri F, et al. Use of tyrosine kinase inhibitors to prevent relapse after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: a position statement of the Acute Leukemia Working Party of the European Society for Blood and marrow transplantation. Cancer. 2016;122(19):2941–51.
Acknowledgements
The authors are grateful to the patient and her family in this study for her collaboration.
Funding
This study was funded by the Sichuan Science and Technology Program, grant no. No.2022YFS0236, and the Clinical Research Program of Sichuan Anti-cancer Association (Qilu), grant no.XH2022-507.
Author information
Authors and Affiliations
Contributions
He GQ and Lei YP wrote the manuscript with contributions from all other authors; Huang DW drafted and revised the paper. Gao J and Yang R performed the topic selection, designed the study and edited the manuscript. All authors contributed to and reviewed the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of West China Second Hospital, Sichuan University. The patient’s parents were informed and consented to participate.
Consent for publication
Written informed consent was obtained from the parents for the publication of any potentially identifiable images or data included in this article.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, Gq., Lei, Yp., Huang, Dw. et al. Philadelphia chromosome-like acute lymphoblastic leukemia with concomitant rearrangements of CRLF2 and ABL1: a pediatric case report. BMC Pediatr 24, 517 (2024). https://doi.org/10.1186/s12887-024-04991-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04991-w