
Abstract
Background
While Miller-Dieker syndrome critical region deletions are well known delineated anomalies, submicroscopic duplications in this region have recently emerged as a new distinctive syndrome. So far, only few cases have been described overlapping 17p13.3 duplications.
Methods
In this study, we report on clinical and cytogenetic characterization of two new cases involving 17p13.3 and 3p26 chromosomal regions in two sisters with familial history of lissencephaly. Fluorescent In Situ Hybridization and array Comparative Genomic Hybridization were performed.
Results
A deletion including the critical region of the Miller-Dieker syndrome of at least 2,9 Mb and a duplication of at least 3,6 Mb on the short arm of chromosome 3 were highlighted in one case. The opposite rearrangements, 17p13.3 duplication and 3p deletion, were observed in the second case. This double chromosomal aberration is the result of an adjacent 1:1 meiotic segregation of a maternal reciprocal translocation t(3,17)(p26.2;p13.3).
Conclusions
17p13.3 and 3p26 deletions have a clear range of phenotypic features while duplications still have an uncertain clinical significance. However, we could suggest that regardless of the type of the rearrangement, the gene dosage and interactions of CNTN4, CNTN6 and CHL1 in the 3p26 and PAFAH1B1, YWHAE in 17p13.3 could result in different clinical spectrums.
Similar content being viewed by others

Background
The diagnosis of human chromosome abnormalities including gain or loss of genomic copy numbers has extremely benefited from the development of advanced molecular cytogenetic methods such as array-CGH. This allows high-resolution pangenomic analysis, in particular in detecting genetic imbalances, defining their size, delimiting translocation breakpoints and analyzing the involved segments [1]. Array-CGH has identified novel co-locating micro-deletions and micro-duplication in the same locus. This has allowed the description of new genomic disorders leading to distinct clinical phenotypes. Recently, the duplication of the entire Miller-Dieker syndrome critical region (MDS) involving PAFAH1B1 and YWHAE genes as well as new co-locating micro-duplications in chromosome 17p13.3 have been defined within duplication syndromes in the MDS locus [2, 3]. Likewise, deletions and duplications of 3p26 region have been described as new emerging syndromes [4,5,6].
In this study, we report a familial translocation (3;17) leading to two different cytogenetic rearrangements resulting in a duplication/deletion of the 17p13.3 critical region for MDS including PAFAH1B1 and YWHAE genes and 3p26 region including CNTN4, CNTN6, CRBN and a part of CHL1. The duplication and deletion of the same chromosomal region resulted as expected in distinct phenotypic features in the offspring.
Methods
Clinical report
Patient1 (the proband)
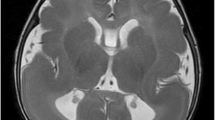
A 2-year-old girl referred for the cytogenetic exploration with a family history of lissencephaly (Fig. 1.II-2), is the second child of a healthy consanguineous Tunisian couple. The patient’s weight at birth was 3500 g (+ 0,6SD). She measured 52 cm (+ 1,05SD) and had a head circumference of 35 cm (+ 0,4SD). At 2 years of age, her height and head circumference were 88 cm (+ 0,9SD) and 45 cm (− 2,5SD), respectively. At physical examination, she had psychomotor development delay and an abnormal behavior including aggressiveness, anger and agitation. Furthermore, she had craniofacial dysmorphic features (Fig. 2a, A’) including a long face, a high forehead, down-slanting palpebral fissures, epicanthus, a wide nose, a long philtrum, a thin upper lip, large and high implanted ears and a pointed chin with micrognathia. In addition, she showed arachnodactyly. Her cerebral magnetic resonance imaging (MRI) was performed at two years and five months of age, and corpus callosum hypoplasia was detected.

Pedigree of the family

Photographs of the patients
Patient 2
The patient (Fig. 1.II-7) presented at 4 months for exploration because of growth retardation, axial hypotonia, seizure and dysmorphic features (Fig. 2b) including a high forehead, a wide nose, low implanted ears and lissencephaly at MRI. She died 10 months later. Her brother (Fig. 1.II-1) suffering from type 1 lissencephaly, also died at an early age.
The proband (II-2) (gray) and her sister (II-7) (striped) carried a der (3) and der (17) respectively. The white triangle and the black diamond represent terminated pregnancies and affected stillborn, respectively.
Karyotype
Metaphase chromosome preparations were obtained by phytohemagglutinin (PHA) stimulated lymphocyte culture according to standard procedures. Chromosome analysis was carried out applying R-banding at a 500-band level according to ISCN 2016 [7] in the patient, parents and sister.
Fluorescent in situ hybridization (FISH)
FISH was performed on blood lymphocytes blocked on metaphases of the patient (II-2), those of her sister (II-7) and those of her mother, according to the standard protocol. One probe screening the chromosome 17 short arm was used: commercial probes; Miller-Dieker/Lissencephaly region probe set: LISI (Red) and RARA (Green) (Vysis) (Abbott Laboratories, IL, USA).
The hybridized chromosomal spreads were analyzed using a fluorescent microscope equipped with appropriate filters and Cytovision FISH system image capture software (Zeiss Axioskop 2 plus). Slides were scored on the basis of the number of probe signals for each metaphase. For each target area ten hybridized metaphases were analyzed.
Array CGH
Oligonucleotide array CGH was performed using the Agilent Human Genome CGH Microarray Kit 44 K®. This microarray consisted of more than 44,000 oligonucleotide probes that spanned both coding and non-coding regions. The coverage of the human genome was made with an average spatial resolution of 75,000 pair bases.
The patient’s DNA as well as a reference DNA was fragmented by heat at 95 °C for 20 min. Each fragmented DNA product was labeled by random priming using either ULS5 or ULS3. After column-purification, probes were denatured and pre-annealed with 5 μg of human Cot-1 DNA, 10 μl of CGH Blocking agent and 55 μl of hybridization buffer. Hybridization was performed at 65 °C during 24 h. The microarray was washed, scanned and analyzed with Agilent Feature Extraction® 9.1 software. Results were interpreted using DNA analytics® 4.5 software. Only imbalances involving three or more adjacent probes were held. The identification of probes with a significant gain or loss was based on the log2 ratio plot deviation from 0 with cutoff values of 0.5 to 1, and − 0.5 to − 1, respectively.
Results
The conventional cytogenetic analysis did not reveal any chromosomal anomalies in the two sisters (II-2/II-7) nor in parents’ karyotypes.
FISH was first performed on the sister (II-7) using the subtelomeric probes (Vysis) of chromosome 17p and showed the absence of a subtelomeric signal on one of the chromosomes 17p (Fig. 3a). This was indicative of a family subtelomeric translocation (Fig. 4). Consequently, using the same probe of chromosome 17p, FISH analysis showed hybridization on the derivative chromosome 3 and on normal chromosome 17 (Fig. 3b), 46,XX.ish t(3;17)(p26.2;p13.3)(LIS1+,subtel3ptel+,subtel3qter+) in the mother. FISH was then performed in the proband (II-2) using 17p probe and showed three signals on the two normal chromosomes 17 and the derivative chromosome 3 (Fig. 3c). This confirmed the duplication of the terminal region of chromosome 17.

FISH analyses. a FISH results from patient II-7 using commercial Miller Dieker/Lissencephaly region probe set: (Lsi LIS1: Red and Lsi RARA: Green) showing the absence of the red fluorescence signal on the arrowed der(17), suggesting that the LIS1 gene is deleted. b FISH results from mother using the same commercial probe, demonstrating the translocation of terminal material from 17p to chromosome 3p (green arrow). c FISH results from patient II-2 using the commercial Miller Dieker/Lissencephaly region probe set showing the presence of three red fluorescence signal on the arrowed der(3) and the two arrowed chr 17, confirming that the LIS1 gene is duplicated

Ideograms of maternal chromosomes 17 and 3 and their derivatives der(17) and der(3)
Ideograms of maternal chromosomes 17 and 3 illustrate the exchange of chromosome material of 17ptel and 3ptel regions due to the reciprocal translocation t(3;17). The patient (II-2) inherited the der(3) mat and the normal paternal chromosomes 17 and 3. The patient (II-7) inherited the der(17) mat and the normal paternal chromosomes 17 and 3.
Aiming to delimit the involved segments, array-CGH analysis was performed on the proband and showed a large deletion of 3,6 Mb on the short arm of chromosome 3, involving 12 OMIM genes and a large duplication of 2,9 Mb on the short arm of chromosome 17, encompassing 61 OMIM genes: 46,XX.arr[GRCh18]3p26.2(224727_3864822)X1,17p13.3(48539_2976723)X3 mat (Fig. 5).

Results of 44 K Agilent oligo array-CGH analysis in patient II-2. A. chromosome 17, showing 17p13.3 duplication of at least 2,9 Mb in size. Chromosome 3, showing 3p26.2 deletion of at least 3,6 Mb in size
Discussion
Adjacent-1 segregation of the translocation t(3;17) in the mother led to two different chromosome imbalances in the children. The first adjacent-1 type gave rise to a derivative 3 (der3) in patient II-2 that resulted in partial monosomy 3p and partial trisomy 17p. On the other hand, the second adjacent-1 type led to a derivative 17 (der17) in patient II-7, thus resulting in partial monosomy 17p and partial trisomy 3p. While deletions of 17p13.3 are associated with a well-known phenotype ranging from Miller Dieker syndrome [3] to partial agenesis of corpus callosum and milder phenotype [8], duplications of the same chromosomal region still need further clinical and molecular characterization. According to the involved genes, 17p13.3 duplications have been divided into either class I or class II leading to different clinical features [2].
So far, to the best of our knowledge, only 13 patients having large 17p13.3 duplications, including the entire MDS comprising both PAFAH1B1 and YWHAE genes have been reported [2, 9,10,11,12,13,14,15] (Fig. 6) with varying sizes and different breakpoints. It has also been reported that these duplications might be the result of parental translocations. They have never involved the 3p26 region.

Schematic illustration of the molecular findings in individuals reported with duplication in the Miller Dieker Syndrome (MDS) Critical Region encompassing both YWHAE and PAFAH1B1 genes
The genomic distances (in base pairs from the 17p telomere) shown at the top of the figure were measured according to ensembl genome browser 59 (hg18). For each patient, a normal copy number is illustrated as a blue line and the duplicated segment as a pink line.
Here, our proband showed a loss of nearly 3,6 Mb on 3p26.2 and a gain of nearly 2,9 Mb on 17p13.3 and shared clinical and dysmorphic features including a high forehead and a triangular chin described in thirteen selected patients with duplication of the MDS region (Table 1). Our patient did not share some of these features whereas he presented arachnodactyly, which is rarely described in patients with partial trisomy of 17p13.3 [2, 9, 11, 16]. The most frequent phenotypic features associated with partial trisomy of 17p13.3 were correlated with the duplication of the PAFAH1B1 and YWAHE genes that were located in the MDS region. It was hypothesized that the duplication of YWHAE might have an effect on neuronal network development and maturation, and was related to mild development delay and facial dysmorphisms while the duplication of PAFAH1B1 that lead to its overexpression, was associated with moderate to severe development delay and structural brain abnormalities [2, 9]. Brain-imaging analysis was performed in seven of the eleven reported patients and only four showed structural brain abnormalities (Table 1), among which Corpus Callosum hypoplasia or agenesis represented the main brain abnormality [9, 10, 13, 14]. Likewise, our patient presented corpus callosum hypoplasia. Curiously, patients reported so far as having the smallest and the largest duplications of the MDS region present normal Magnetic Resonance Imaging (MRI) (P1/ [10]; P1/ [15]). This suggests that this heterogeneity depends on the size of the duplication and the involved genes as well as on the involvement of other gene interactions and modifier genes. Indeed, it has been proven that transgenic mice with increased lis1 expression in the developing brain revealed abnormalities in the neuroepithelium such as the thinning of the ventricular zone, and the ectopic positioning of mitotic cells [9]. Furthermore, lis1 overexpression affected both radial and tangential migration with a migration delay in radial migration at E13.5 and tangential migration at E12.5 rather than E14.5 [10]. However, subtelomeric neuronal migration defects are not expected to be detected by MRI scans [9]. Consequently, we can postulate that the overexpression of LIS1 gene could account for the phenotype of our patient particularly corpus callosum hypoplasia.
Numerous features in this case might be attributed to genes that are lost in chromosome 3p in addition to 17p13.3 duplication as a result of adjacent-1 malsegregation of the maternal balanced translocation. In fact, it has been shown that terminal 3p deletions are responsible for a rare contiguous gene disorder (OMIM# 613792) [17]. Interestingly, we reviewed six previously reported cases having 3p deletion, compared them to the present case report, and noted that the most frequent features are microcephaly, corpus callosum hypoplasia and facial dysmorphia [18, 19] (Table 2). Conversely, some studies reported cases with 3p deletion and normal phenotypes [17, 22, 23]. In other studies, the authors have even hypothesized that distal 3p deletion is probably associated with normal intelligence and normal physical features [18, 24] and that the severity of the phenotype depends on the size of the deletion as well as on the gene content and the disrupted genes involved in the breakpoints, essentially CNTN4, CNTN6 and CRBN [25, 26]. The CNTN6 gene plays a crucial role in the development, maintenance, and plasticity of functional neuronal networks in the central nervous system. It has been shown that Cntn6 deficiency in mice causes profound motor coordination abnormalities and learning difficulties [25]. Owing to its function, we suggest that CNTN6 gene could be responsible for the observed psychomotor development retardation in the current case. On the other hand, CNTN4 is known to be involved in axon growth, guidance, and fasciculation [25] and it probably contributes to the behavioral abnormalities in our patient showing aggressiveness, anger and agitation. In fact, cntn4 knockout mice showed morphological, neurological and behavioral abnormalities [25]. The deletion also included CRBN gene that plays a crucial role in brain development [26]. In fact, CRBN protein is part of the DCX protein ligase complex involved in the regulation of the surface expression of certain types of ion channels in neuronal memory synapses. Furthermore, 3p26 deletion disrupted a more distal gene: CHL1 that plays a crucial role in the development of the cortex by regulating neuronal differentiation and axon guidance [27]. Previous studies suggested CHL1 as a dosage-sensitive gene with a major role in intellectual disabilities [28]. Interestingly, Frints hypothesized that a reduction equal to 50% of chl1 in the developing brain marked cognitive deficit [29].
Interestingly, both 3p deletion and17p duplication could share the same network in neuronal migration since both anomalies lead to corpus callosum hypoplasia and pachygyria. So far, both PAFAH1B1genes duplicated in 17p and CNTN6 as well as CRBN genes deleted in 3p affected the process of cortical development by destabilization of microtubules and alteration of axon growth and axon guidance [25, 30, 31].
Neuronal migration is a complex process that involves several actors and factors in order to elaborate an appropriate cell migration from the ventricular zone into the cortical plate during normal brain development [32]. Mutations and chromosomal aberrations can alter chromosome 3D organization. This alteration could play a more important role than we believe it does in the chromosomal interactions and transcriptional regulation of genes. In fact, it has been shown that chromatin 3D modification could disturb the topologically associating domains (TADs) and consequently the regulation of gene expression [33]. Such alteration could explain the phenotypic variability in human disease ranging from a milder phenotype to a microdeletion/microduplication syndrome. Furthermore, this variability can be explained by the consanguinity in this family, which reduces the suitability of individuals by increasing the degree of homozygosity and promoting the development of deleterious recessive genes [34]. Finally, patients carrying CNVs known to have broad variable clinical expressivity and possibly incomplete penetrance, may benefit from whole exome sequencing analysis in the near future.
Conclusions
The variability of genes, which are mapped in the involved regions (3p and 17p), and the description of the clinical characteristics of our patient contribute to the confirmation and further delineation of the associated characteristics to the partial trisomy of 17p13.3 encompassing the entire MDS critical region as well as the partial monosomy of chromosome 3p26.2. Various genes and structural chromosomal anomalies have been discovered as being involved in this process. However, the exact molecular basis of brain malformations still needs further studies.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the [ArrayExpress] repository. [https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-8748].
Abbreviations
- Array CGH:
-
Array comparative genomic hybridization
- CHL1 :
-
close homolog of L1
- CNTN4 :
-
contactin 4
- CNTN6 :
-
contactin 6
- CRBN :
-
cereblon
- ISCN:
-
International System for Human Cytogenetic Nomenclature
- OMIM:
-
Online Mendelian Inheritance in Man
- PAFAH1B1 :
-
platelet activating factor acetylhydrolase 1b regulatory subunit 1
- SD:
-
standard deviation
- YWHAE :
-
tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein epsilon
References
Sanlaville D, Lapierre JM, Coquin A, Turleau C, Vermeesch J, Colleaux L, Borck G, Vekemans M, Aurias A, Romana SP. La CGH microarray: Principe et applications en pathologie constitutionnelle. Archives de Pediatrie. 2005;12(10):1515–20.
Bruno DL, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, Martin CL, Douglas J, Nowak C, Adam MP, Kooy RF, Van der Aa N, Reyniers E, Vandeweyer G, Stolte-Dijkstra I, Dijkhuizen T, Yeung A, Delatycki M, Borgström B, Thelin L, Cardoso C, van Bon B, Pfundt R, de Vries BB, Wallin A, Amor DJ, James PA, Slater HR, Schoumans J. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47(5):299–311.
Blazejewski SM, Bennison SA, Smith TH, Toyo-Oka K. Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3. Frontiers in Genetics. 2018;9(80).
Cargile CB, Goh DLM, Goodman BK, Chen XN, Korenberg JR, Semenza G, Thomas GH. Molecular cytogenetic characterization of a subtle interstitial del(3)(p25.3p26.2) in a patient with deletion 3p syndrome. Am J Med Genet. 2002;109(2):133–8.
Chen CP, Huang MC, Chern SR, Kuo YL, Chen YN, Wu PS, Chen LF, Pan CW, Wang W. Distal 3p duplication and terminal 7q deletion associated with nuchal edema and cyclopia in a fetus and a review of the literature. Taiwan J Obstet Gynecol. 2015;54(3):297–302.
Kaur A, Khetarpal S. 3p deletion syndrome. Indian Pediatr. 2013;50(8):795–6.
McGowan-Jordan J, Simons A, Schmid M. An international system for human cytogenomic nomenclature. Cytogenet Genome Res. 2016;149:1–2.
Hannachi H, Mougou-Zerelli S, BenAbdallah I, Mama N, Hamdi I, Labalme A, Elghezal H, Sanlaville D, Saad A. Clinical and molecular characterization of a combined 17p13.3 microdeletion with partial Monosomy 21q21.3 in a 26-year-old man. Cytogenetic and Genome Research. 2011;135(2):102–10.
Bi W, Sapir T, Shchelochkov OA, Zhang F, Withers MA, Hunter JV, Levy T, Shinder V, Peiffer DA, Gunderson KL, Nezarati MM, Shotts VA, Amato SS, Savage SK, Harris DJ, Day-Salvatore DL, Horner M, Lu XY, Sahoo T, Yanagawa Y, Beaudet AL, Cheung SW, Martinez S, Lupski JR, Reiner O. Increased LIS1 expression affects human and mouse brain development. Nat Genet. 2009;41(2):168–77.
Roos L, Jønch AE, Kjaergaard S, Taudorf K, Simonsen H, Hamborg-Petersen B, Brøndum-Nielsen K, Kirchhoff M. A new microduplication syndrome encompassing the region of the miller-Dieker (17p13 deletion) syndrome. J Med Genet. 2009;46(10):703–10.
Hyon C, Marlin S, Chantot-Bastaraud S, Mabboux P, Beaujard MP, Al Ageeli E, Vazquez MP, Picard A, Siffroi JP, Portnoï MF. A new 17p13.3 microduplication including the PAFAH1B1 and YWHAE genes resulting from an unbalanced X;17 translocation. European Journal of Medical Genetics. 2011;54(3):287–91.
Kiiski K, Roovere T, Zordania R, Von Koskull H, Horelli-Kuitunen N. Prenatal diagnosis of 17p13.1p13.3 duplication. Case Rep Med. 2012;2012:1–5.
Ruiz Esparza-Garrido R, Velzquez-Wong AC, Araujo-Sols MA, Huicochea-Montiel JC, Velzquez-Flores MÁ, Salamanca-Gmez F, Arenas-Aranda DJ. Duplication of the miller-Dieker critical region in a patient with a subtelomeric unbalanced translocation t(10;17)(p15.3;p13.3). Molecular Syndromology. 2012;3(2):82–8.
Curry CJ, Rosenfeld JA, Grant E, Gripp KW, Anderson C, Aylsworth AS, Saad TB, Chizhikov VV, Dybose G, Fagerberg C, Falco M, Fels C, Fichera M, Graakjaer J, Greco D, Hair J, Hopkins E, Huggins M, Ladda R, Li C, Moeschler J, Nowaczyk MJ, Ozmore JR, Reitano S, Romano C, Roos L, Schnur RE, Sell S, Suwannarat P, Svaneby D, Szybowska M, Tarnopolsky M, Tervo R, Tsai AC, Tucker M, Vallee S, Wheeler FC, Zand DJ, Barkovich AJ, Aradhya S, Shaffer LG, Dobyns WB. The duplication 17p13.3 phenotype: Analysis of 21 families delineates developmental, behavioral and brain abnormalities, and rare variant phenotypes. American Journal of Medical Genetics, Part A. 2013;161(8):1833–52.
Kucharczyk M, Jezela-Stanek A, Gieruszczak-Bialek D, Kugaudo M, Cieslikowska A, Pelc M, Krajewska-Walasek M. Oculocutaneous albinism in a patient with 17p13.2-pter duplication– a review on the molecular syndromology of 17p13 duplication. Biomedical Papers. 2015;159(2):333–7.
Primerano A, Colao E, Villella C, Nocera MD, Ciambrone A, Luciano E, D'Antona L, Vismara MFM, Loddo S, Novelli A, Perrotti N, Malatesta P. A cryptic balanced translocation (5;17), a puzzle revealed through a critical evaluation of the pedigree and a FISH focused on candidate loci suggested by the phenotype. Mol Cytogenet. 2015;8:70.
Moghadasi S, van Haeringen A, Langendonck L, Gijsbers ACJ, Ruivenkamp CA. A terminal 3p26.3 deletion is not associated with dysmorphic features and intellectual disability in a four-generation family. American Journal of Medical Genetics Part A 9999. 2014(11);1–6.
Cuoco C, Ronchetto P, Gimelli S, Béna F, Divizia MT, Lerone M, Mirabelli-Badenier M, Mascaretti M, Gimelli G. Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet Journal of Rare Diseases. 2011;6:12.
Chen CP, ChenYY CSR, Wu PS, Su JW, Chen WL, Wang W. Prenatal diagnosis of a distal 3p deletion associated with fetoplacental chromosomal discrepancy and confined placental mosaicism detected by array comparative genomic hybridization. Taiwanese Journal of Obstetrics and Gynecology. 2013;52(2):278–84.
Ben-Abdallah-Bouhjar I, Hannachi H, Labalme A, Gmidène A, Mougou S, Soyah N, Gribaa M, Sanlaville D, Elghezal H, Saad A. Chromosomal microarray analysis of functional xq27-qterdisomy and deletion 3p26.3 in a boy with Prader-Willi like features and hypotonia. European Journal of Medical Genetics. 2012;55(8–9):461–5.
Kashevarova AA, Nazarenko LP, Schultz-Pedersen S, Skryabin NA, Salyukova OA, Chechetkina NN, Tolmacheva EN, Rudko AA, Magini P, Graziano C, Romeo G, Joss S, Tümer Z, Lebedev IN. Single gene microdeletions and microduplication of 3p26.3 in three unrelated families: CNTN6 as a new candidate gene for intellectual disability. Mol Cytogenet. 2014;7(1):1–10.
Shrimpton AE, Jensen KA, Hoo JJ. Karyotype–Phenotype Analysis and Molecular Delineation of a 3p26 Deletion/8q24.3 Duplication Case With a Virtually Normal Phenotype and Mild Cognitive Deficit. American Journal of Medical Genetics, 140A. 2006;388–91.
Gijsbers CJ, van Haeringen A, Bosch CAJ, Hansson K, Verschuren M, Bakker E, Breuning MH, Ruivenkamp CA. A subtle familial translocation t(3;21) (p26.3;q22.3): an apparently healthy boy with a 3p deletion and 21q duplication. Cytogenet Genome Res. 2010;128(4):245–9.
Pohjola P, de Leeuw N, Penttinen M, Kääriäinen H. Terminal 3p deletions in two families-correlation between molecular karyotype and phenotype. Am J Med Genet A. 2010;152A(2):441–6.
Zuko A, Kleijer KTE, Oguro-Ando A, Kas MJH, Van Daalen E, Van Der Zwaag B, Burbach JP. Contactins in the neurobiology of autism. Eur J Pharmacol. 2013;719(1–3):63–74.
Higgins JJ, Tal AL, Sun X, Hauck SCR, Hao J, Kosofosky BE, Rajadhyaksha AM. Temporal and spatial mouse brain expression of Cereblon, an Ionic Channel regulator involved in human intelligence. J Neurogenet. 2010;24(1):18–26.
Katic J, Loers G, Kleene R, Karl N, Schmidt C, Buck F, Zmijewski JW, Jakovcevski I, Preissner KT, Schachner M. Interaction of the cell adhesion molecule CHL1 with vitronectin, integrins and the plasminogen activator inhibitor-2 promotes CHL1-induced neurite outgrowth and neuronal migration. J Neurosci. 2014;34(44):14606–23.
Li C, Liu C, Zhou B, Hu C, Xu X. Novel microduplication of CHL1 gene in a patient with autism spectrum disorder: a case reportand a brief literature review. Mol Cytogenet. 2016;9:51.
Frints SGM, Marynen P, Hartmann D, Fryns JP, Steyaert J, Schachner M, Rolf B, Craessaerts K, Snellinx A, Hollanders K, D'Hooge R, De Deyn PP, Froyen G. CALL interrupted in a patient with non-specific mental retardation: gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet. 2003;12(13):1463–74.
Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nature Cell Biol. 2000;2(11):767–75.
Papuc SM, Hackmann K, Andrieux J, Vincent-Delorme C, Budisteanu M, Arghir A, Schrock E, Ţuţulan-Cuniţă AC, Di Donato N. Microduplications of 3p26.3p26.2 containing CRBN gene in patients with intellectual disability and behavior abnormalities. European Journal of Medical Genetics. 2015;58(5):319–23.
Guerrini R, Dobyns W. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;13(7):710–26.
Scott F Gilbert. Developmental Biology, 6th edition. 2000. https://www.ncbi.nlm.nih.gov/books/NBK9983/. Acessed 2000.
Solignac M, Periquet G, Anxolabehere D, Petit C. Génétique et Evolution 1: La variation, les gènes dans les populations. Ed des Sciences et des Arts. Acessed: Herman; 1995.
Acknowledgements
We are very grateful to the family members for their kind participation and for their continuous interest in this study. We also thank the scientific and technical team of the cytogenetics Department at Farhat Hached University Teaching Hospital (Sousse, Tunisia) and Ms. N. Kerkni for English editing.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SMZ contributed to conception and design. MHA and SD contributed to all experimental work, analysis and interpretation of data. HH contributed partially to the experiment and analysis work. AT, KBH and AM referred the patients and assured medical monitoring. SMZ and SD were responsible for the consultation. SMZ and AS were responsible for overall supervision. MHA and WS drafted the manuscript, which was revised by SMZ. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the local Ethics Board of the University Teaching Hospital Farhat Hached. Written informed consent to participate in this study was obtained from the parents.
Consent for publication
Written informed consent was obtained from the parents for photo and clinical data publication.
Competing interests
All the authors have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hadj Amor, M., Dimassi, S., Taj, A. et al. Neuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype. BMC Med Genet 21, 26 (2020). https://doi.org/10.1186/s12881-020-0966-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-0966-9