
Abstract
Background
Microcephaly is a disorder characterized by severe impairment in brain development, reduced brain and head size. Congenital severe microcephaly is very rare, and NDE1 deletion and genetic mutations are important contributors.
Case presentation
Single nucleotide polymorphism (SNP) chromosomal microarray analysis (CMA) and muation screening of NDE1 gene were performed in an 8-month patient with severe congenital microcephaly, and/or his parents. Genetic studies found a 16p13.11 deletion containing NDE1 gene, and a novel NDE1 mutation c.555_556GC > CT on the non-deleted homolog, inherited from his phenotypically normal parents, respectively. The 2 bp nucleotide change results in a missense mutation p.K185 N and a nonsense mutation p.Q186X. We also conducted literaturte review to compare the clinical phenotypes of our patient to those of cases previously reported with NDE1 mutations, and found all patients had mental retardation, severe microcephaly, and corpus callosum agenesis.
Conclusion
This is the first Chinese reported with microcephaly caused by NDE1 mutations. NDE1 is a critical pathogenetic gene in severe congenital microcephaly. Sequencing NDE1 and CMA in patients with severe congenital microcephaly may be warranted.
Similar content being viewed by others

Background
The 16p13.11 locus is a genomic hotspot especially rich in low-copy repeats. These highly homologous DNA sequences are more likely to form copy number mutations through non-allelic homologous recombination [1]. The deletions of 16p13.11 are associated with a wide spectrum of disorders including schizophrenia, autism, mental retardation, intellectual disability, epilepsy, and mild microcephaly [2,3,4,5,6]. The 16p13.11 region contains a core set of genes including NDE1, the most important candidate gene for the neurodevelopmental phenotypes associated with the 16p13.11 microdeletions. NDE1 encodes the nuclear distribution factor E-homolog 1 (nudE) protein that localizes to the centrosome and mitotic spindle poles, interacting with the cytoplasmic dynein complex and Lissencephaly-1 (LIS1) [7, 8]. This protein plays a crucial role in microtubule organization, mitosis and neuronal migration, essential for the process of mammalian encephalisation and human cerebral cortex growth [9].
To date, several cases of autosomal recessive extreme microcephaly caused by NDE1 mutations have been reported. Herein we report that a patient, with severe congenital microcephaly, had 16p13.11 deletion containing NDE1 gene combined with a novel NDE1 mutation on the non-deleted homolog. We also compared the clinical phenotypes of all patients so far reported with NDE1 mutations.
Case presentation
The CARE guidelines were followed in this case. The patient is an 8 months old male that had been diagnosed with extreme microcephaly. The patient was the first child of healthy, non-consanguineous parents, delivered after a 37 weeks gestation by cesarean section. The mother had a spontaneous abortion history after a 12 weeks gestation. The patient displayed microcephaly at birth but the physical parameters were not available. At the age of 8 months, physical examination showed severe microcephaly (Fig. 1a) with head circumference was 32 cm, 9.6 SD below the mean, marked hypertonia on his extremities, and global developmental delay. Magnetic resonance imaging scans showed the agenesis of the corpus callosum, enlargement of the posterior lateral ventricles, and a simplified gyral pattern of the cerebral cortex with scarce and wide gyri in parietal and occipital lobes (Fig. 1b). Electroencephalography showed slow background activity without epileptiform discharge.

The typical phenotype and laboratory results. The patient at 8 months of age showed severe microcephaly (a). MRI showed the agenesis of the corpus callosum, enlargement of the posterior lateral ventricles, and a simplified gyral pattern of the cerebral cortex (b). The Diagram of chromosome 16 and the 1.22 M microdeletion on 16p13.11 in the patient detected by CMA (c and d). The NDE1 sequence chromatogram of the patient and his parents, and arrows show the 2 base-pairs changes (e)
Informed consent was obtained from the parents and the study protocols were reviewed and approved by the medical ethics committee of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science & Technology. Genomic DNA was extracted from peripheral blood using E.Z.N.A. ® MicroElute Genomic DNA Kit (Omega Bio-tek) from the patient and his parents. The samples from the patient and his parents were studied by the InfiniumOmniZhongHua-8 DNA Analysis BeadChip (Illumina), using for CNVs analysis larger than 30 Kb. CMA found that the patient had a 1.22 Mb deletion (hg19:15,070,916–16,292,323) on 16p13.11 inherited from his phenotypically normal mother (Fig. 1d). The deleted region contains 20 genes, including ABCC1, ABCC6, FOPNL, MPV17L, MYH11, NDE1 and NTAN1. This boy had severe congenital microcephaly resembling that described in those patients with homozygous mutations in NDE1 gene [9,10,11,12], so we attempted to detect genetic mutations in NDE1 by performing Sanger Sequencing. The NDE1 coding exons and flanking intronic sequences of the samples were amplified by PCR and then sequenced by Sanger sequencing using standard methods. Sequences were compared with normal reference sequences for NDE1 (NCBI reference number NM_001143979.1, NP_001137451.1). The analysis identified a c.555_556GC > CT mutation in NDE1 on the non-deleted homolog, inherited from his phenotypically normal father (Fig. 1e). The 2 bp nucleotide change results in a missense mutation (p.K185 N) and a stop codon (p.Q186X).
Discussion and conclusion
We reported an 8-month patient with severe microcephaly, who harbored maternal 16p13.11 deletion combined with a paternal NDE1 mutation. To date, 14 patients from 8 families with microcephaly involved NDE1 gene have been reported, and these patients manifested severe congenital microcephaly, profound intellectual disability, and early onset seizure [9,10,11,12].
Microcephaly is a cortical malformation disorder characterized by severe impairment in brain development, small brain size, and a substantial reduction in neuronal number. Several genes have been identified to cause this condition, such as ASPM, WDR62, CDK5RAP2, CENPJ, SIL, and BRIT1 [13,14,15], and these genes are involved in proliferation of the progenitor cells by affecting centrosome, mitotic spindle assembly, and cell cycle progression. NDE1-associated microcephaly is much more severe than the forms caused by defects in these genes, and patients with NDE1 mutations often exhibit neuronal lamination defects, such as a simplified gyral structure or lissencephaly [9,10,11,12], and NDE1 might have multiple roles in neocortical development.
Table 1 showed a comparison of the clinical findings of our patient with those cases reported previously [9,10,11,12]. All patients had mental retardation, severe microcephaly, and corpus callosum agenesis. Ventricular enlargement was presented in all patients except that Paciorkowski et al. did not describe this issue in their study. All but our patient displayed hypoplastic cerebellum or a proportionate reduction in the size of cerebellum, and epilepsy occurred in most of patients. Currently, it is unclear why the cerebellum of our patient normally appeared in brain MRI. These two mutations in our case, being inherited from their healthy heterozygous parents, were predicted to produce truncated proteins or deleted allele. As previously reported, immunoblot assay did not show detectable NDE1 protein in patient lymphoblast and, whereas the heterozygous parents showed ~50% reduction in protein level, suggesting the instability of mutant protein and subsequent degradation [10].
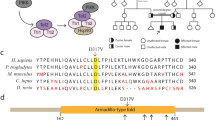
NDE1 consists of 10 exons and a consensus start codon is located within exon 3 (NM_001143979.1) (Fig. 2a). Human NDE1 is a protein with 335 amino acids, harboring self-association domain, LIS1 interaction domain, and the C-terminal conserved domain (Fig. 2b). The C-terminal domain is essential for centrosomal localization of NDE1 and for its interaction with CENP-F (centromere protein F) and dynein [16,17,18]. LIS1 is essential for neural progenitor division and cortical migration, and its genetic mutations cause Miller-Dieker syndrome [19, 20]. CENP-F, a nuclear matrix component localizing at kinetochores during mitosis, is required for kinetochore localization of NDE1, LIS1, and Dynein [18]. Cytoplasmic dynein is a multisubunit complex involved in a variety of cellular functions, including mitotic spindle control, checkpoint regulation, transport of organelles, and directed nuclear and cell migration [16, 17, 21]. The corrected localization and activation of the dynein complex require the presence of many adaptors, such as CENP-F, LIS1, and NDE1. All these proteins work together to link spindle microtubes to kinetochores, being responsible for mitotic cell division. Recent studies have found that NDE1 is required in cell cycle progression and postmitotic neuronal migration, and NDE1 inhibition caused substantial numbers of radial glia progenitor cells’ nuclei to become arrested by impairing primary cilium dynamics, apical nuclear migration, and entry into mitosis [22]. All these findings could explain the defects in NDE1 contribute to extreme micrpcephaly and other neurodevelopmental conditions.

The diagram of human NDE1 (NM_001143979.1). The coding sequences are shown in black and all the reported mutations are labeled (a). The predicted NDE1 protein derived from the mutations (b)
To date, several mutations in NDE1 have been reported. Bakircioglu et al. and Alkuraya et al. reported three distinct homozygous frameshift mutations of NDE1, c.83 + 1G > T (p.A29QfsX114), c.684_685delAC (p.P229WfsX85) and c.733dupC (p.L245PfsX70) [9, 10]. The c.83 + 1G > T leads to the formation of a novel splice site and to a frameshift after NDE1 exon 3, and the novel open reading frame would be predicted to maintain the N-terminal 28 native amino acids followed 113 novel amino acids [9]. The mutant protein completely lost the conserved C-terminal domain, including interacting domains with CENP-F and dynein, and the centrosomal localization domain, the majority of LIS1 binding domain as well [9]. The c.684_685delAC resulted in a premature stop codon at position 313, and the truncated protein lost the highly conserved C-terminal CENP-F and dynein interaction domain, and also failed to present at the centrosome. However, the mutant protein would be predicted to interact with LIS1 [9]. The c.733dupC resulted in a truncated protein with 244 native amino acids of the wild-type protein on the N terminus followed by 69 abnormal amino acids. The consequence of this mutation is similar to that of c.684_685delAC, which have been proved in vitro studies [9, 10], and these truncated mutant proteins failed to bind to the dynein complex and to localize properly to the centrosome. Guven et al. identified a homozygous deletion that encompasses NDE1 exon 3 containing the initiation codon, predicted to result in a null allele [11]. Paciorkowski et al. describe two patients with inherited deletions of 16p13.11 encompassing the entire NDE1 gene on one allele combined with a nonsense mutation (p.R44X) or a frameshift mutation (p.R303TfsX11) in the non-deleted NDE1 [12]. Our patient had the deletion of the entire NDE1 gene on one allele combined with a c.555_556GC > CT on the non-deleted homolog, resulting in a missense mutation (p.K185 N) and a nonsense mutation (p.Q186X). The nonsense mutation is predicted to cause protein truncation in the C-terminal domain of NDE1. If stable, the truncated protein may fail to localize properly to the centrosome due to the loss of domains interacting with CENP-F and dynein. This heterozygote of c.555G > C (p.K185 N) and c.556C > T was found to be only presented in one case in East Asian among in populations with more than 120,000 individuls, including East asian, African, Latino, European, etc. in genome aggregation database (http://gnomad.broadinstitute.org/), respectively. There was no corresponding literature about these two variants, and it is unclear whether they were presented in one person. There is no report in the first Asian Diploid genome (http://yh.genomics.org.cn/).
In, conclusion, we reported a NDE1 genetic mutation combined with 16q13.11 microdeletion in a boy with extreme microcephaly. To our knowledge, this is the first Chinese reported with microcephaly caused by NDE1 mutations. Our findings enrich the understanding of NDE1 mutations and the associated phenotypes. It is warranted to perform mutation screening in NDE1 gene for patients with severe micropcephaly.
Nomenclature
Nucleotide symbol
-
A adenosine
-
C cytidine
-
G guanosine
-
T thymidine
-
U uridine
Amino acid residue symbol
-
A Alanine
-
K Lysine
-
L Leucine
-
N Asparagine
-
P Proline
-
Q Glutamine
-
R Arginine
-
T Threonine
-
W Tryptophan
-
X stop codon
Abbreviations
- ABCC1:
-
ATP binding cassette subfamily C member 1
- ABCC6:
-
ATP binding cassette subfamily C member 6
- ASPM:
-
abnormal spindle microtubule assembly
- bp:
-
base pairs
- BRIT1:
-
BRCA1 C terminus repeat inhibitor of hTERT
- CDK5RAP2:
-
CDK5 regulatory subunit associated protein 2
- CENP-F:
-
centromere protein F
- CENPJ:
-
centromere protein J
- CMA:
-
Chromosomal Microarray Analysis
- CNVs:
-
copy number variations
- del:
-
deletion
- dup:
-
duplication
- FOPNL:
-
FGFR1OP N-terminal like
- fs:
-
frameshift
- Kb:
-
kilobase(s) or 1000 bp
- LIS1:
-
Lissencephaly-1
- MPV17L:
-
MPV17 mitochondrial inner membrane protein like
- MYH11:
-
myosin heavy chain 11
- NDE1:
-
nuclear distribution factor E-homolog 1
- NTAN1:
-
N-terminal asparagine amidase
- PCR:
-
Polymerase Chain Reaction
- SD:
-
standard deviation
- SIL:
-
STIL, centriolar assembly protein
- WDR62:
-
WD repeat domain 62
References
Martin J, Han C, Gordon LA, Terry A, Prabhakar S, She X, et al. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004;432(7020):988–94.
de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133(Pt 1):23–32.
Heinzen EL, Radtke RA, Urban TJ, Cavalleri GL, Depondt C. Need AC, et al. rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86(5):707–18.
Ingason A, Rujescu D, Cichon S, Sigurdsson E, Sigmundsson T, Pietilainen OP, et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 2011;16(1):17–25.
Ramalingam A, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY, et al. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56(7):541–4.
Ullmann R, Turner G, Kirchhoff M, Chen W, Tonge B, Rosenberg C, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28(7):674–82.
Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44(2):279–93.
McKenney RJ, Vershinin M, Kunwar A, Vallee RB, Gross SP. LIS1 and NudE induce a persistent dynein force-producing state. Cell. 2010;141(2):304–14.
Bakircioglu M, Carvalho OP, Khurshid M, Cox JJ, Tuysuz B, Barak T, et al. The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet. 2011;88(5):523–35.
Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, et al. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet. 2011;88(5):536–47.
Guven A, Gunduz A, Bozoglu TM, Yalcinkaya C, Tolun A. Novel NDE1 homozygous mutation resulting in microhydranencephaly and not microlyssencephaly. Neurogenetics. 2012;13(3):189–94.
Paciorkowski AR, Keppler-Noreuil K, Robinson L, Sullivan C, Sajan S, Christian SL, et al. Deletion 16p13.11 uncovers NDE1 mutations on the non-deleted homolog and extends the spectrum of severe microcephaly to include fetal brain disruption. Am J Med Genet A. 2013;161a(7):1523–1530.
TW Y, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, et al. Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat Genet. 2010;42(11):1015–20.
Bilguvar K, Ozturk AK, Louvi A, Kwan KY, Choi M, Tatli B, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467(7312):207–10.
Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome? Trends Genet. 2009;25(11):501–10.
Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, et al. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28(3):681–96.
Stehman SA, Chen Y, McKenney RJ, Vallee RB. NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J Cell Biol. 2007;178(4):583–94.
Vergnolle MA, Taylor SS. Cenp-F links kinetochores to Ndel1/Nde1/Lis1/dynein microtubule motor complexes. Curr Biol. 2007;17(13):1173–9.
Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, et al. Isolation of a miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364(6439):717–21.
Coquelle FM, Caspi M, Cordelieres FP, Dompierre JP, Dujardin DL, Koifman C, et al. LIS1, CLIP-170's key to the dynein/dynactin pathway. Mol Cell Biol. 2002;22(9):3089–102.
Hirohashi Y, Wang Q, Liu Q, Li B, Du X, Zhang H, et al. Centrosomal proteins Nde1 and Su48 form a complex regulated by phosphorylation. Oncogene. 2006;25(45):6048–55.
Doobin DJ, Kemal S, Dantas TJ, Vallee RB. Severe NDE1-mediated microcephaly results from neural progenitor cell cycle arrests at multiple specific stages. Nat Commun. 2016;7(12551.
Acknowledgements
We gratefully acknowledge the kind cooperation and agreement of patient and his parents. We also thank Dr. Jun Jiang, Jun Lin, and Nannan Zheng for their generous help and when we prepared our case report.
Funding
This research was supported in part by Wuhan Creative Platform of Clinical Medical Research for Neurological Diseases in Children (2014–160), Wuhan Yellow Crane (Medicine and Healthcare) (2016–01), Health and Family Planning Commission of Hubei Province(WJ2015MB247), Health and Family Planning Commission of Wuhan (WX16C13).
Availability of data and materials
All data and material are available from one of corresponding authors, Xuelian He, who can be contacted by email on the email address (hexuelian2013@hotmail.com).
Author information
Authors and Affiliations
Contributions
Performed experiment: LT, PZ, XC. Conceived and designed the experiments: XH, JS. Acquisition and analyzed the data: PZ, CW, BB. Contributed reagents/materials/analysis tools: CW, XC. Clinical data, including patient’s image: JS, BB. Wrote the paper: LT, XH, BB. Final approval of the publication: JS, BB. All authors have read and approved the manuscript to be published.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained from the parents and the study protocols were approved by the medical ethics committee of Wuhan Children’s Hospital.
Consent for publication
Written consent was obtained from the parents of patient for the publication, including child’s data and images.
Competing interests
The author(s) declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tan, L., Bi, B., Zhao, P. et al. Severe congenital microcephaly with 16p13.11 microdeletion combined with NDE1 mutation, a case report and literature review. BMC Med Genet 18, 141 (2017). https://doi.org/10.1186/s12881-017-0501-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0501-9