
Abstract
Background
Bardet Biedl Syndrome (BBS) is a rare condition of multi-organ dysfunction with characteristic clinical features of retinal degeneration, truncal obesity, postaxial polydactyly, genital anomaly, intellectual disability and renal dysfunction. It is a hetero-genetic disorder and nineteen BBS genes have been discovered so far.
Methods
Whole genome SNP genotyping was performed by using CytoScan® 750 K array (Affymetrix). Subsequently, the segregation of the disease locus in the whole family was carried out by genotyping STS markers within the homozygous interval. Finally, the mutation analysis was performed by Sanger DNA sequencing.
Results
In the present molecular study a consanguineous Pakistani family, with autosomal recessive BBS, was analyzed. The clinical analysis of affected individuals presented with synpolydactyly, obesity, intellectual disability, renal abnormality and retinitis pigmentosa. The presented phenotype was consistent with the major features of BBS syndrome. Homozygosity mapping identified a common homozygous interval within the known BBS9 locus. Sequence analysis of BBS9/PTHB1 gene revealed a single base deletion of c.299delC (p.Ser100Leufs*24) in exon 4. This frame-shift mutation presumably leads to a 122 amino acid truncated protein with complete loss of its C-terminal PTHB1 domain in combination with a partial loss of the N-terminal PTHB1 domain as well. BBS9/PTHB1 gene mutations have been shown to be associated with BBS syndrome and to the best of our knowledge this study reports the first Pakistani family linked to the BBS9 gene.
Conclusion
Our molecular findings expand the mutational spectrum of BBS9 gene and also explain the genetic heterogeneity of Pakistan families with BBS syndrome. The growing number of mutations in BBS genes in combination with a detailed phenotypical description of patients will be helpful for genotype-phenotype correlation, targeted genetic diagnosis, prenatal screening and carrier testing of familial and non-familial BBS patients.
Similar content being viewed by others


Background
Bardet Beidl syndrome (BBS) is a multi-symptomatic ciliopathy. The major clinical manifestations of this disorder are cone-rod dystrophy, truncal obesity, postaxial polydactyly, genital anomaly, learning disability and renal dysfunction [1]. Clinically BBS is a heterogeneous disorder with variable inter- and intra-familial features [2], therefore its diagnosis is established by the presence of four or three of the major features and two minor features like speech delay, brachydactyly/syndactyly, cardiovascular anomalies etc. [3, 4]. The incidence rate of BBS varies among the different ethnicities, and occur with the ratio of 1: 156,000 in Tunisia [5], 1: 140,000–160,000 in North America to Europe, 1: 17,000 in Kuwait, and 1: 18,000 in Newfoundland [6, 7].
Genetically BBS is segregated in an autosomal recessive fashion with high degree of locus and allele heterogeneity. The combinatorial approach of SNP microarray (for homozygosity mapping) with targeted exome sequencing (for mutation analysis) has speed-up the discovery of causative gene and its underlying pathogenic mutation [8, 9]. Genetic analysis of BBS patients have identified nineteen disease responsible genes (BBS1-19), most of which encode BBSome protein (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8/TTC8, BBS9/PTHB1, BBS17/LZTFL1 and BBS18/BBIP1) while some produce basal body interacting protein (BSS13/MKS1, BBS14/EP290/NPHP6, BBS15/WDPCP and BBS16/SDCCAG8), chaperonin complex protein (MKKS, BBS10 and BBS12), E3 ubiquitin ligase (BBS11/TRIM32), and a GTPase protein complex (BBS3/ARL6 and BBS19/IFT27) [10, 11]. Physiologically BBSome is of prime importance in ciliogenesis due to its functional role in membrane trafficking in primary cilium [12]. The mutational spectrum of BBS genes is variable among different populations. A high proportion of European and Caucasian families are reported to have mutation in BBS1 and BBS10 gene with the ratio of 23.2 % and 20 % respectively [13, 14], while in the Arab population most of the mutations are found in BBS1, BBS2, BBS3, BBS4 and BBS8 [10, 15–17] with a total prevalence rate of 67 %. Although no prevalence data of the BBS genes have been estimated yet in the Pakistani population however most of the mutations occur in BBS1 [18], BBS3/ARL6 [19], BBS10 [19, 20], and BBS12 [21].
In the present linkage study, we have ascertained a Saraiki origin BBS family from the Kirikhaisor village of D.I.Khan in the KPK province of Pakistan. It was a consanguineous family with two affected siblings presenting with BBS features. Homozygosity mapping in combination with Sanger sequencing of a candidate gene identified a novel single base deletion mutation in BBS9 gene [(c.299delC) & (p.Ser100Leufs*24)]. This frame shift mutation presumably leads to truncated nonfunctional protein. These data present the first report of the BBS9 gene mutation in a Pakistani family and thus increase the mutational spectrum of Bardet Beidl syndrome in this ethnic region.
Methods
From sampling to DNA isolation
The consanguineous family for the present molecular investigation was ascertained from the rural area of D.I.Khan in the Khyber-Pakhtoonkhwa Province of Pakistan. The study was approved by the ethical review board of Gomal Centre of Biochemistry and Biotechnology, Gomal University D.I.Khan, Pakistan. Moreover, informed written consent was obtained from all individuals participating in the study. The family was visited at its residence and blood samples were collected. The patients were examined in detail for all apparent phenotypic features to document the major as well as the minor BBS symptoms. The parents were also counseled in detail on prenatal, perinatal, and neonatal medical problems. Subsiquently, the DNA was isolated from the peripheral blood sample by using standard Phenol-Chloroform method.
From genome to gene analysis
To investigate the underlying disease gene mutation, genome-wide SNP genotyping was performed by using Affymetrix CytoScan 750 K SNP microarray. The SNP data were analyzed using the Chromosome Analysis Suite (ChAS) version 1.2 (Affymetrix) to identify the homozygous by descend region (HBD). The perfect segregation of identified disease associated HBD region in the complete family was determined by genotyping STR markers D7S493 (36.08 cM), D7S516 (42.93 cM), D7S484 (53.73 cM), and D7S510 (59.8 cM) {Rutgers map B37 [22]} within the homozygous region. For statistical significance of linkage, two point and multi point LOD scores were calculated by using Superlink and GeneHunter software packages plugged-in the EasyLinkage plus v5.02 software. The analysis was performed assuming a disease allele frequency of 0.001 with full penetrance at zero recombination fraction [23].
The primers for protein coding exons of BBS9 gene were designed using the online version of Primer 3 plus software (version 0.4.0) [24]. The sequence reactions were performed with BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystem, USA) and the sequencing products were analyzed by the Genetic analyzer 3130xl (Life Technologies, USA). The sequence data were then aligned with the BBS9 reference sequence via blast like alignment tool (BLAT) tool package of UCSC Genome Browser [25].
Results
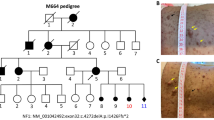
In this study, four individuals from a multi-generation family were enrolled for molecular investigation of an underlying disease gene mutation. The recruited individuals consist of both affected siblings (IV-4 and IV-5), one unaffected brother (IV-6) and an asymptomatic carrier mother (III-2). Pedigree analysis revealed an autosomal recessive mode of disease segregation (see Fig. 1).

Family pedigree and facial photographs of patient IV-4 and IV-5. Symbols, tagged with asterisks, indicate individuals selected for genetic analysis in addition to both affected persons
Clinical findings
The clinical examination of both BBS patients demonstrated bilateral postaxial synpolydactyly of hands and feet, mild progressive retinal degeneration, intellectual disability, renal dysfunction, and obesity as a major BBS features, while mild hearing loss was also noticed as a minor feature in both affected individuals. The pattern of postaxial synpolydactyly in the female showed hepta-dactyly of left hand and foot but the right hand and foot had six digits, while the affected male had only hepta-dactyly of the right hand. The detailed clinical description of both affected individuals is shown in Table 1.
Genetic findings
Whole genome SNP genotyping and linkage analysis
Genome-wide homozygosity mapping revealed a 18.45 Mb (HG19: chr7: 21,596,876-39,320,676) long HBD region between markers rs56356943 to rs12112084, on the short arm of chromosome 7. Segregation mapping by STR markers in the complete family, through STR markers, confirmed the inheritance of the homozygous region with disease etiology. Linkage analysis generated a suggestive two point and multipoint LOD scores of 0.92 and 1.26 respectively at same marker D7S484 (53.73 cM) with zero recombination fraction. The identified homozygous interval harbored the BBS9 gene which is reported to be associated with BBS.
Sequence analysis of the BBS9 gene
Sanger DNA sequencing of the BBS9 gene revealed a homozygous deletion of cytosine nucleotide in exon 4 (c.299delC). At protein level the reported deletion is predicted to result in a frame-shift mutation p.Ser100Leufs*24 (UniProtKB accession: Q3SYG4). The predicted premature stop codon presumably disrupts the open reading frame and results in a truncated polypeptide of 122 amino acids with complete loss of C-terminal PTHB1 domain (see Fig. 2). The identified sequence variant is not present in exome variant server (URL: http://exac.broadinstitute.org/), exome aggregation consortium (URL: http://evs.gs.washington.edu/EVS/), The Singapore Human Mutation and Polymorphism database (URL: http://shmpd.bii.a-star.edu.sg/) the Indian Genetic Disease database (URL: http://www.igdd.iicb.res.in/IGDD/home.aspx) and 100 normal controls from Pakistan (n = 50) and Arab population (n = 50).

a- Autozygosity image, showing the common homozygous interval on 7p14.3 that harbors the known BBS9 gene and the defect was detected in its exon 4. b- Sequence chromatogram of BBS9 exon 4 is showing a homozygous deletion of cytosine in an affected individual (IV-4), heterozygous mutation in a carrier (III-2) and wild-type sequence in an normal individual (IV-6). c- The protein truncating mutation is depicting a loss of complete PTHB1 C-terminus domain along with some part of N-terminus, leaving behind a 122 amino acid truncated protein
Discussion
Ciliopathies are a distinct group of genetic disorders which are caused by primary ciliary dysfunction with pleiotropic effect. Bardet Biedl syndrome is a rare autosomal recessive ciliopathic disorder characterized by retinal degeneration, intellectual disability, obesity, renal complications and polydactyly as predominant phenotypes. It is a heterogenetic disorder with 19 causative genes, whose protein products together constitute centrosome and/or basal body which is physiologically involved in ciliary function [26, 27]. Here in this report, we discovered a novel single base deletion in the 4th exon of BBS9 gene (c.299delC), which presumably leads to truncated protein with complete loss of the C terminal PTHB1 domain (UniProtKB accession:Q3SYG4). The presence of the premature stop codon will likely lead to degradation of the resulting mRNA through Nonsense-mediated mRNA Decay.
BBS9 protein is the component of BBSome protein complex, a central entity of ciliogenesis, which is localized in the primary cilia and peri-centriolar region. Ten BBSome subunits (from BBS1 to 10) have been discovered, and BBS9 has a possible role in associating other subunits [12]. STRING protein interaction database (URL: www.string-db.org) predicted strong interaction of BBS9 with BBS5 protein, which suggests that BBS5 could be involved in the assembly of BBSome subunits through BBS9 interaction. Hence, loss of function mutation in BBS9 may affect the integrity of BBSome proteins complex due to a structural abnormality in cilia. This structural aberration could be the reason for disassembly of BBSome subunits due to defective BBS9-BBS5 protein interaction which might be required for proper organization. The knockdown studies in zebrafish and mouse have clearly demonstrated the significant role of BBS9 in cilia biogenesis [28]. Although extensive work has been performed for the genetic screening of BBS genes, but the actual function of most BBS proteins are yet to be discovered.
The mutational spectrum of BBS9 gene has reported 12 missense/non-sense mutations, 2 splice site changes, 2 small deletions, 2 small insertions and 4 gross deletions in BBS phenotype (HGMD database; URL: http://www.hgmd.cf.ac.uk/ac/index.php), where as one complex rearrangement in BBS9 is associated with Wilms tumor (HGMD database; [29]). The clinical spectrum of both patients reported in this study is consistent with previous reports, except for a few novel phenotypic variations. These clinical variations included digestion problems (constipation) in both affected (IV-4 & IV-5), irregular menstrual cycle (IV-5), strabismus (IV-4), and synpolydactyly of internal digits (IV-4 & IV-5).
The molecular genetics testing of any inherited disorder either involves multi-gene panel or the most implicated genes. In case of BBS, the high cost of multi-gene panels (BBS1- BBS19) and weakly established genotype-phenotype correlations favor the screening of highly prevalent BBS1 and BBS10 genes. Hence, keeping in view the genetic counseling of BBS patients, the large number of reported BBS genes and unclear phenotypic association data, genetic screening of the most implicated BBS1 and BBS10 is considered as cost effective. Nevertheless, prevalences of supposedly rare BBS genes such as BBS9 might vary in different ethicities and therefore prioritization of genes have to be adapted accordingly in a diagnostic setup.
In summary, our data represent the first BBS9 gene mutation being reported in a Pakistani family associated with Bardet Biedl syndrome. In addition to cardinal features of BBS, slight clinical variations were also observed in Pakistani siblings. It is assumed that the current study will contribute to the elucidation of the genetic heterogeneity of BBS patients and that the screening of additional BBS cases in future might detect the mutational hotspot and thus would be helpful in designing a multi-gene panel for routine diagnosis.
Conclusion
Here in this study we report the first Pakistani family segregating a novel deletion mutation in BBS9 gene (c.299delC). The mutation shifts the reading frame and presumably results in a truncated protein with loss of functional PTHB1 domains (p.Ser100Leufs*24). The current finding has increased the mutational spectrum of the Bardet Biedl Syndrome mutation in the Pakistani population and thereby will increase our knowledge to understand the potential role of the BBS9 protein in BBSome protein complex.
Abbreviations
- BBS:
-
Bardet Biedl syndrome
- D.I.Khan:
-
Dera Ismail Khan
- KPK:
-
Khyber-Pakhtoonkhwa
- DNA:
-
Deoxy ribonucleic acid
- SNP:
-
Single nucleotide polymorphism
- ChAS:
-
Chromosome analysis suite
- HBD:
-
Homozygous by Descend
- cM:
-
Centi Morgan
- LOD:
-
Logarithm of odd
- BLAT:
-
Blast like alignment tool
- Mb:
-
Mega base
- STR:
-
Short tandem repeat
- Ser:
-
Serine
- Leu:
-
Leucine
- fs:
-
Frame shift
- HGMD:
-
Human gene mutation database
References
M’hamdi O, Ouertani I, Chaabouni-Bouhamed H. Update on the genetics of bardet-biedl syndrome. Mol Syndromol. 2014;5(2):51–6.
Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–37.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–46.
Beales PL, Katsanis N, Lewis RA, Ansley SJ, Elcioglu N, Raza J, et al. Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am J Hum Genet. 2001;68:606–16.
M’hamdi O, Ouertani I, Maazoul F, Chaabouni H. Prevalence of Bardet-Biedl syndrome in Tunisia. J Community Genet. 2011;2:97–9.
Farag TI, Teebi AS. High incidence of Bardet-Biedl syndrome among the Bedouin. Clin Genet. 1989;36:463–4.
Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: a 22 year prospective, population-based, cohort study. Am J Med Genet A. 2005;132:352–60.
Redin C, Le Gras S, Mhamdi O, Geoffroy V, Stoetzel C, Vincent MC, et al. Targeted high-throughput sequencing for molecular diagnosis of genetically heterogeneous diseases: efficient mutation detection in Bardet-Biedel and Alström syndromes. J Med Genet. 2012;49:502–12.
Scheidecker S, Etard C, Pierce NW, Geoffroy V, Schaefer E, Muller J, et al. Exome sequencing of Bardet- Biedl syndrome patient identifies a null mutation in the BBSome subunit BBIP1 (BBS18). J Med Genet. 2014;51(2):132–6.
M’hamdi O, Redin C, Stoetzel C, Ouertani I, Chaabouni M, Maazoul F, et al. Clinical and genetic characterization of Bardet-Biedl syndrome in Tunisia: defining a strategy for molecular diagnosis. Clin Genet. 2014;85(2):172–7.
Aldahmesh MA, Li Y, Alhashem A, Anazi S, Alkuraya H, Hashem M, et al. IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum Mol Genet. 2014;23(12):3307–15.
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–13.
Forsythe E, Beales PL. Eur J Hum Genet. 2013;21:8–13.
Janssen S, Ramaswami G, Davis EE, Hurd T, Airik R, Kasanuki JM, et al. Mutations analysis in Bardet-Biedl syndrome by DNA pooling and massively parallel resequencing in 105 individuals. Hum Genet. 2011;129:79–90.
Smaoui N, Chaabouni M, Sergeev YV, Kallel H, Li S, Mahfoudh N, et al. Screening of the eight BBS genes in Tunisian families: no evidence of triallelism. Invest Ophthalmol Vis Sci. 2006;47:3487–95.
Abu Safieh L, Aldahmesh MA, Shamseldin H, Hashem M, Shaheen R, Alkuraya H, et al. Clinical and molecular characterization of Bardet-Biedl syndrome in consanguineous populations: The power of homozygosity mapping. J Med Genet. 2010;47:236–41.
Abu Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, et al. In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet. 2012;20:420–7.
Ajmal M, Khan MI, Neveling K, Tayyab A, Jaffar S, Sadeque A, et al. Exome sequencing identifies a novel and a recurrent BBS1 mutation in Pakistani families with Bardet-Biedlsyndrome. Mol Vis. 2013;19:644–53.
Khan S, Ullah I, Irfanullah, Touseef M, Basit S, Khan MN, et al. Novel homozygous mutations in the genes ARL6 and BBS10 underlying Bardet-Biedl syndrome. Gene. 2013;515(1):84–8.
Agha Z, Iqbal Z, Azam M, Hoefsloot LH, van Bokhoven H, Qamar R. A novel homozygous 10 nucleotide deletion in BBS10 causes Bardet-Biedl syndrome in a Pakistani family. Gene. 2013;519(1):177–81.
Pawlik B, Mir A, Iqbal H, Li Y, Nürnberg G, Becker C, et al. A Novel Familial BBS12 Mutation Associated with a Mild Phenotype: Implications for Clinical and Molecular Diagnostics Strategies. Mol Syndromol. 2010;1(1):27–34.
Matise TC, Chen F, Chen W, De La Vega FM, Hansen M, He C, et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–6.
Lindner TH, Hoffmann K. easyLINKAGE: A PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics. 2005;21:405–7.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86.
Kent WJ. BLAT - the BLAST-like alignment tool. Genome Res. 2002;12:656–64.
Andrade LJ, Andrade R, França CS, Bittencourt AV. Pigmentary retinopathy due to Bardet-Biedl syndrome: case report and literature review. Arq Bras Oftalmol. 2009;72(5):694–6.
Hemachandar R. Bardet-Biedl syndrome: A rare cause of end stage renal disease. Int J Appl Basic Med Res. 2015;5(1):70–2.
Veleri S, Bishop K, Dalle Nogare DE, English MA, Foskett TJ, Chitnis A, et al. Knockdown of Bardet-Biedl Syndrome Gene BBS9/PTHB1 Leads to Cilia Defects. PLoS One. 2012;7(3), e34389.
Vernon EG, Malik K, Reynolds P, Powlesland R, Dallosso AR, Jackson S, et al. The parathyroid hormone-responsive B1 gene is interrupted by a t(1;7)(q42;p15) breakpoint associated with Wilms’ tumour. Oncogene. 2003;22(9):1371–80.
Electronic References
1- UniProtKB URL: http://www.uniprot.org.
2- String Database (Known and predicted protein-protein interactions) URL: http://string-db.org/.
3- Human Gene Mutation Database URL: http://www.hgmd.cf.ac.uk/ac/index.php.
4- exome variant server URL: http://exac.broadinstitute.org/.
5- exome aggregation consortium URL: http://evs.gs.washington.edu/EVS/.
6- The Singapore Human Mutation and Polymorphism database URL: http://shmpd.bii.a-star.edu.sg/.
7- Indian Genetic Disease database URL: http://www.igdd.iicb.res.in/IGDD/home.aspx.
Acknowledgements
Special thanks to all volunteer family members for participating in the present study. MAK is extremely grateful to the Higher Education Commission of Pakistan for granting funds as a startup research grant (M-IPFP/HRD/HEC/2011/346) for molecular screening of neurological disorders. MAK appreciates the valuable contribution of his nephew “Abdul Ahad Khan Gandapur” and his friend Pir Jalal in recruiting this family for the study. Many thanks to Maria Langer-Winter for critical reading and correcting the manuscript. MAK is grateful to Sulman Basit, Assistant Professor, Taibah University Almadinah Almunawarah, Medina, KSA, for screening of the identified novel variant (c.299delC) in his in-house database of normal controls.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MAK was involved in recruiting the BBS family, clinical analysis of data, genetic analysis of data, and manuscript drafting. SM performed genetic mapping and mutation screening. MZ carried out family recruitment, blood sampling, DNA isolation, and clinical analysis. CW supervised the whole study, helped in data analysis, edited and refined the manuscript. Finally, all authors have read and approved the refined manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Khan, M.A., Mohan, S., Zubair, M. et al. Homozygosity mapping identified a novel protein truncating mutation (p.Ser100Leufs*24) of the BBS9 gene in a consanguineous Pakistani family with Bardet Biedl syndrome. BMC Med Genet 17, 10 (2016). https://doi.org/10.1186/s12881-016-0271-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-016-0271-9