
Abstract
Background
NPAS3 encodes a transcription factor which has been associated with multiple human psychiatric and neurodevelopmental disorders. In mice, deletion of Npas3 was found to cause alterations in neurodevelopment, as well as a marked reduction in neurogenesis in the adult mouse hippocampus. This neurogenic deficit, alongside the reduction in cortical interneuron number, likely contributes to the behavioral and cognitive alterations observed in Npas3 knockout mice. Although loss of Npas3 has been found to affect proliferation and apoptosis, the molecular function of NPAS3 is largely uncharacterized outside of predictions based on its high homology to bHLH–PAS transcription factors. Here we set out to characterize NPAS3 as a transcription factor, and to confirm whether NPAS3 acts as predicted for a Class 1 bHLH–PAS family member.
Results
Through these studies we have experimentally demonstrated that NPAS3 behaves as a true transcription factor, capable of gene regulation through direct association with DNA. NPAS3 and ARNT are confirmed to directly interact in human cells through both bHLH and PAS dimerization domains. The C-terminus of NPAS3 was found to contain a functional transactivation domain. Further, the NPAS3::ARNT heterodimer was shown to directly regulate the expression of VGF and TXNIP through binding of their proximal promoters. Finally, we assessed the effects of three human variants of NPAS3 on gene regulatory function and do not observe significant deficits.
Conclusions
NPAS3 is a true transcription factor capable of regulating expression of target genes through their promoters by directly cooperating with ARNT. The tested human variants of NPAS3 require further characterization to identify their effects on NPAS3 expression and function in the individuals that carry them. These data enhance our understanding of the molecular function of NPAS3 and the mechanism by which it contributes to normal and abnormal neurodevelopment and neural function.
Similar content being viewed by others

Background
NPAS3 [Neuronal PAS (period-ARNT-single minded)-domain containing 3] encodes a transcription factor of the basic Helix Loop Helix–PAS (bHLH–PAS) family expressed in the developing central nervous system [1, 2]. NPAS3 was originally characterized in humans as the causative locus of intellectual disability and psychosis in a Scottish family, as it was broken by a reciprocal translocation that segregated with disorder [1, 3]. Since its discovery as a potential “schizophrenia gene”, NPAS3 has been robustly associated with neurodevelopmental and neuropsychiatric disorders commonly characterized by alterations in white matter connectivity, and intellectual disability. Large scale deletions including NPAS3 have been associated with holoprosencephaly, holoprosencephaly microform and other gross neurodevelopmental abnormalities [4,5,6]. Smaller deletions physically limited to NPAS3 have been reported as associated with intellectual impairment and disorders of psychosis [7, 8]. Genome-wide studies have identified NPAS3 as associated with bipolar disorder and schizophrenia [9,10,11]. Single nucleotide variation affecting the coding regions of NPAS3 have been associated with neuropsychiatric disorders, including schizophrenia [12, 13]. These data are strongly suggestive of a role for NPAS3 in normal neurodevelopment and neuropsychological function.
The effect of loss of Npas3 has been studied using mouse knockout models which have identified deficits in neurodevelopment resulting in altered neuroanatomy, as well as an almost complete loss of adult neurogenesis in the dentate gyrus of the hippocampus [14, 15]. Disruption Npas3 expression was found to contribute to behavioral deficits which include hallmarks of hippocampal dysfunction, including reduced performance on tasks dependent on hippocampal memory, as well as altered emotional tone [14, 15]. Deletion of Npas3 was found not to result in reduced proliferation of neuroprogenitors in the dentate gyrus of the hippocampus, but instead in increased markers of apoptosis [16]. During development, Npas3 deletion results in reduced formation of cortical interneurons born in the subpallial ganglionic eminences [17]. As such, NPAS3 appears to be critical for neurogenic processes, with potentially far-reaching effects.
Since its discovery, NPAS3 has been characterized as a bHLH–PAS transcription factor based on predicted functional domains [2, 18]. bHLH–PAS proteins contain a bHLH DNA binding and protein interaction domain, followed by PAS domains, two degenerate repeat 70 aa domains, which are involved in protein interaction and ligand binding [19, 20]. A transactivation domain or repressive domain may be encoded C-terminal to the bHLH and PAS domains. All bHLH–PAS proteins are thought to act as heterodimers, requiring interaction with a general heterodimeric partner, such as ARNT, to create a functional heterodimer capable of regulation of target genes [19]. These heterodimers interact through residues in both the bHLH and PAS domains, where the PAS domains specify the interaction partner, while the bHLH domains are able to homo- and heterodimerize with other bHLH-containing proteins [21].
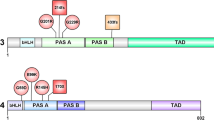
Our understanding of the molecular mechanism of gene regulatory function driven by NPAS3 is lacking. Aryl hydrocarbon receptor nuclear translocator (ARNT) has long been considered to be the obligate heterodimeric partner of NPAS3, however, the interaction between NPAS3 and ARNT had not been molecularly assessed until recent studies of mouse Npas3 and Arnt [22, 23]. NPAS3 and ARNT have been shown to cooperatively regulate genes involved in fibroblast growth factor (FGF) and sonic hedgehog (SHH) signaling, however whether this cooperation involved physical interaction was not demonstrated [24]. Microarray studies in human cells identified hundreds of target genes differentially regulated by expression of NPAS3, however, which are direct targets was not assessed [25]. Of the genes identified in this study, VGF (non-acronymic) has been shown to be regulated by NPAS3 in a manner dependent on constructs proximal to the promoter region [25, 26]. Neither physical association with the promoter region, nor the contribution of ARNT to this regulation were assessed. Recent ChIP-seq (chromatin immunoprecipitation next generation sequencing) studies have identified multiple targets of Npas3 which are differentially regulated in the mouse hippocampus, however the mechanism by which Npas3 regulates these genes is unknown [27]. In this study we performed experiments to characterize the functional domains of NPAS3 in protein interaction and gene-regulatory function. We assessed the relative contribution of the bHLH, PAS and C-terminal putative transactivation domains to these functions. Furthermore, we generated three variants previously identified in the human population, including two psychiatric illness-associated variants p.Val304Ile (c.910G>A, rs146677388) and p.Ala552Pro (c.1654G>C, rs12434716), and one rare population variant, p.Gly697Ser (c.2089G>A, rs141427321), for effects on NPAS3 function.
Methods
Plasmids
The coding sequence of NPAS3 transcript variants 1 (NM_001164849.1 encoding isoform 1 NP_001158221.1, 933 aa) and 2 (NM_022123.1 encoding isoform 2 NP_071406.1, 901 aa) were purchased from Origene and cloned using the Gateway subcloning system (Invitrogen) into a Gateway converted pcI-HA (hemagglutinin tagged) vector. pHTN-CMV-neo (Promega) was also converted to the Gateway system and NPAS3 was subcloned into it to generate a HaloTag construct. HaloTag-ARNT transcript variant 6 (NM_001286036.1 encoding isoform 6 NP_001272965.1 with a silent substitution of c.768G>C, p.Val188 = that is not predicted to change the protein) was purchased (Promega), and pcDNA3.1-ARNT transcript variant 1 (NM_001668.3 encoding isoform 1 NP_001659.1) was acquired from DNASU [28]. Domain constructs of NPAS3 were generated from transcript variant 1 to isolate the bHLH domain (residues 1–125), PAS domains (residues 116–450) and C-terminus (residues 451–933) (Fig. 1a). Domains were cloned into a Gateway converted pcI-HA to generate N-terminally HA-tagged constructs.

NPAS3 and ARNT interact through their bHLH and PAS domains. a Ideogram of NPAS3 constructs used. Exon numbering relative to NPAS3 transcript variant 1 corresponding to NPAS3 isoform 1 (933 aa). Only coding regions of exons are depicted. Domain constructs and variant numbering are relative to isoform 1. NPAS3 isoform 2 (901 aa) was also characterized. Red bHLH domain, Blue PAS domains, Green TAD (C-terminal transactivation domain). b Western blots of HaloTag pull-downs demonstrating that all tested isoforms of NPAS3 and ARNT are able to robustly interact, which is not observed with the empty HaloTag vector. Bands in the top panel of pull-down lane of ‘prey’ protein, indicating physical association of HA-NPAS3 isoforms with the HaloTag-ARNT construct. Middle and bottom panels are results for the HaloTag-clone and HaloTag-empty constructs. As HaloTag constructs are covalently linked to the resin, they will be depleted in the pull-down sample, demonstrating functionality of the HaloTag. Molecular weight of HaloTag-ARNT, 121 kDa, HaloTag construct with no cDNA: 34 kDa, Full-length NPAS3: 101 kDa. c Reciprocal HaloTag pull down data using HaloTag-NPAS3 constructs and untagged ARNT isoform 1. Molecular weight of HaloTag-NPAS3, 135 kDa. d HaloTag-NPAS3 pull-down of endogenously expressed ARNT. All pull-downs have been performed at least twice
Variants tested were generated through site directed mutagenesis of the clone of NPAS3 transcript variant 1 in the Gateway pDONR221 vector (Invitrogen). The c.1654G>C (p.Ala552Pro) and c.2089G>A (p.Gly697Ser) variants were generated using the KapaHiFi Hot Start kit (Kapa Biosystems) and the following primers: (NPAS3-G1654C-F 5′-CGGTGCTCTGGGCCCGATGCAGATCAA-3′, NPAS3-G1654C-R 5′-TTGATCTGCATCGGGCCCAGAGCACCG-3′, G2089A-F 5′-CCCGCAGGGCAGCGGCGGTGG-3′, NPAS3-G2089A-R 5′-CCACCGCCGCTGCCCTGCGGG-3′, variant nucleotides underlined). The c.910G>A (p.Val304Ile) variant was generated using a synthesized 504 bp gene block (IDT DNA) between the AvrII and XhoI (both NEB) sites in the NPAS3 clone, containing the indicated variant. The variant was assembled into digested pDONR221-NPAS3 transcript variant 1 using the Gibson Assembly Cloning Kit (NEB), and recombined into the pcI-HA destination vector.
Cell culture and transfections
HEK 293T cells were purchased from ATCC and cultured in Dulbecco’s Modified Eagle Medium (DMEM) with high glucose (Sigma) in a 37 °C incubator with a humidified 5% CO2 atmosphere. Cells were subcultured at a ratio of 1:5 to 1:10 every 2–3 days, once they reached 75% confluence. For experiments, cells were plated at 2.2 × 106 cells per 10 cm plate and allowed to recover for 24 h before transfection. Transfections were performed using Mirus TransIT LT-1 or Mirus TransIT Express (LT-1 replacement, MirusBio) per the manufacturer’s protocol. Briefly, 4 μg of total transfected DNA was used per reaction, with 12 μl of transfection reagent in 500 μl of serum-free media. Transfection reactions were mixed and incubated for 30 min at room temperature and added dropwise to cells. Cells were harvested 48 h after transfection.
Immunoblot and protein::protein interaction studies
Transfected HEK 293T cells were washed twice and collected by centrifugation at 800×g in PBS (phosphate buffered saline) and frozen to enhance lysis. Cells were thawed, lysed with Mammalian Lysis Buffer (Promega) supplemented with protease inhibitor cocktail (Promega) and 1 mM PMSF (phenylmethylsulfonyl fluoride) per the manufacturer’s protocol. Samples were kept on ice during purification. Insoluble material was precipitated by centrifugation at 10,000×g for 5 min at 4 °C. For western blotting, samples were quantified using the BioRad Protein assay (BioRad) and spectrophotometry at 600 nm and 50 μg of protein was run per sample. Pull down reactions were performed using the HaloTag Mammalian Pull-Down protocol (Promega) per manufacturer’s instructions. Samples were reserved for analysis of the input, flow-through and pull-down samples.
Protein lysates and HaloTag pull-down samples were electrophoresed on 8–15% polyacrylamide gels at 140 V until the dye front reached the end of the gel. Proteins were transferred to nitrocellulose membranes using wet transfer with Towbin buffer. Transfers were run at 30 V overnight at 4 °C. Blots were rinsed with water and blocked for 1 h in LI-COR Block (LI-COR), before being probed with primary and secondary antibodies for 1 h each, with four washes in PBS-Tween buffer after each antibody. Blots were probed with the following antibodies: αHaloTag (mouse, Promega G921A) at 1/10 000, αNPAS3 (guinea pig, generated by Pocono Rabbit Farms & Laboratory to custom oligopeptide C-terminal to the PAS domain, validation shown in Additional file 1: Figure S1) at 1/10 000, αARNT (rabbit, Cell Signaling Technologies D28F3), donkey anti rabbit AlexaFluor 680 (Life Technologies A10043) at 1/25 000, goat anti-guinea pig IRDye 800 (Rockland 606 132 129) at 1/25 000, donkey anti mouse AlexaFluor 680 (Life Technologies A10038) at 1/25 000. Probed blots were scanned using the LI-COR Odyssey scanner using default scan parameters, and processed using LI-COR Image Studio software.
Immunofluorescence
HEK 293T were plated in 6-well plates with sterile coverslips at 3 × 105 cells per well and allowed to recover for 24 h before being transfected with 1 μg of DNA and 3 μl of Mirus TransIT LT1 (MirusBio). Forty-eight hours after transfection, cells were fixed in 2% paraformaldehyde buffered in PBS for 20 min, washed twice in PBS 0.05% Triton-X100 (PBSX) and blocked in PBSX 5% BSA for 15 min. Coverslips were probed with primary antibodies αHA (mouse, Santa Cruz sc-7392, 1/500) and αARNT (rabbit, Cell Signaling Technologies D28F3, 1/250) for 1 h. Coverslips were washed twice and probed with donkey anti-mouse AlexaFluor594 (Invitrogen A21203, 1/1000) and donkey anti-rabbit AlexaFluor488 (Invitrogen A21206, 1/1000) for 1 h. Coverslips were washed twice and stained with DAPI (4′,6-diamidino-2-phenylindole, 2 μg/ml) for 5 min before mounting onto slides with ProLong Antifade Gold reagent (Invitrogen). Slides were visualized using a Leica DM RE fluorescent microscope and images processed using Northern Elite Eclipse (EMPIX), and ImageJ [29] to add scale bars.
Identification of potential co-targets of NPAS3 and ARNT
The genes identified as differentially regulated by NPAS3 in a previous microarray study were cross-referenced to the Encyclopedia of DNA Elements (ENCODE) ChIP-seq data for ARNT in K562 cells (experiment ID ENCSR155KHM) visualized in the UCSC genome browser [25, 30, 31]. Potential co-targets were selected for screening based on the presence of peaks in both replicates and pooled analysis of the ENCODE ChIP-seq study, as well as peak identification by conservative and optimal peak calling algorithms, resulting in a score out of 5. VGF was also selected for assessment due to deeper characterization in the index microarray study [25].
Gene expression analysis
HEK 293T cells were harvested 48 h post-transfection using the RNeasy mini kit (QIAGEN) per the manufacturer’s protocol. RNA samples were quantified by Nanodrop and 500 ng was used as input into the Quantitect RT kit (QIAGEN) for cDNA synthesis per the protocol. qPCR was performed on 0.5 μl of the resultant cDNA per reaction using the KAPA SYBR FAST Universal 2X qPCR master mix (KapaBioscience) per the manufacturer protocol, with 1 μl of 2 μM primers per reaction. PCR reactions were performed in triplicate. Cycling was performed in a CFX96 Touch (BioRad) with parameters as follows: 3 min initial denaturation at 95 °C, followed by 40 cycles of 5 s denaturation at 95 °C with 25 s extension at 60 °C. All qPCR data were normalized to three housekeeping genes: hydroxymethylbilane synthase (HMBS), hypoxanthine phosphoribosyltransferase 1 (HPRT1) and succinate dehydrogenase complex flavoprotein subunit A (SDHA). Primers used are listed in Additional file 2: Table S1.
HaloCHIP
HEK 293T cells were plated and transfected with HaloTag NPAS3 isoform 1 with and without ARNT isoform 1, or HaloTag-ARNT isoform 6 with and without NPAS3 isoform 1 as described above. Forty-eight hours after transfection, a representative plate was counted, and 1 × 107 cells were input per reaction. Cells were fixed for 10 min in 1% formaldehyde and cross-linking was quenched with 0.125 M glycine for 5 min. Cells were washed twice with PBS and collected in 1 mL PBS. Lysis was performed per the HaloCHIP protocol with the optional cytoplasmic lysis step and homogenization using a Dounce homogenizer and 25 passes of the B-pestle. Chromatin was sheared by sonication and benzonase digestion based on the protocol outlined in [32], using four cycles of sonication with a Biodisrupter probe sonicator at medium high (60%) intensity for 30 s on/off. MgCl2 was added to a final concentration of 1 mM followed by incubation for 15 min with 250 U of benzonase per reaction to result in shearing to 1 kb or smaller in size. ethylenediaminetetraacetic acid (EDTA) pH 8.0 was added to 5 mM to terminate the reaction. An input sample of 1% was reserved, and HaloCHIP was performed per the manufacturer’s instructions with slight modification. Blocked lysates were obtained by incubation of nuclear lysates with the HaloCHIP blocking reagent, a dye which is catalyzed by the HaloTag enzyme, terminally inhibiting its catalytic function, resulting in tagged constructs being unable to covalently bind the resin. The resin was washed with 2 ml of each wash indicated in the HaloCHIP protocol, including the optional high salt wash buffer. The first three washes included 5 mM EDTA pH 8.0 to ensure the benzonase was inactive during washing. Samples were eluted overnight in kit elution buffer supplemented with 5 mM EDTA pH 8.0 at 65 °C. DNA was purified using QIAquick gel extraction kit (QIAGEN) per the manufacturer’s protocol.
Enrichment was assessed using endpoint PCR and gel electrophoresis. From each ChIP sample, 1 μl was added to a master mix of the GoTaq green reaction buffer with 10 μM primers. Primers used are listed in Additional file 2: Table S2. Cycling conditions were as follows: denaturation at 95 °C for 2 min, 30 cycles of 95 °C for 30 s, 60 °C for 15 s, 72 °C for 15 s; amplification was completed by a 5 min incubation at 72 °C. PCR reactions were run on a 1.5% agarose 1X TBE (Tris borate EDTA) gel, stained with ethidium bromide, and visualized on a UV transilluminator.
Reporter gene assay
Promoter constructs were generated synthetically using sequences designed on the UCSC genome browser human genome build hg19 [33]. Regions were selected to include HaloCHIP positive regions in our studies, ENCODE ARNT ChIP-seq peaks in the same interval, as well as binding sites for ARNT homo- and heterodimeric complexes predicted by ConTra v3.0 [34]. Binding sites for Hypoxia Inducible Factor 1 Alpha (HIF1A)::ARNT (MA0259.1), ARNT (MA0004.1) and Aryl Hydrocarbon Receptor (AhR)::ARNT (MA0006.1) were predicted in the 1000 bp proximal promoter, 5′UTR and intron 1 of VGF and TXNIP (thioredoxin interacting protein) using position-weight matrices from the JASPAR database indicated above, using the ConTra v3.0 settings: stringency core = 0.95, and similarity matrix = 0.85. For VGF, the promoter construct consisted of 797 bp upstream of the transcription start site as defined by NM_003378.3, as well as 13 bp of the first exon, as indexed in UCSC genome browser human genome build hg19. This region was synthesized (IDT DNA) for assembly into pGL4.10 linearized using XhoI and HindIII (both NEB) with the Gibson Assembly kit (NEB). For TXNIP, the region starting 867 bp upstream of the transcription start site defined by NM_006472.5, as well as 68 bp of exon 1, was cloned as described for VGF.
For luciferase, HEK 293T cells were plated at 5 × 104 cells per well of a 24-well plate. Cells were plated and transfected in triplicate per condition. Cells were transfected using Mirus TransIT LT1 or TransIT Express (MirusBio) per the manufacturer’s protocol and 50 ng of pGL4.10- promoter-firefly luciferase, 150 ng of each driver construct, 0.1 ng pGL4.7-TK-renilla luciferase. Drivers were pcI-HA-NPAS3 isoform 1 and pcDNA3.1-ARNT isoform 1 or empty vectors (pcI-HA and pcDNA4). Forty-eight hours after transfection, cells were processed per the Dual-Luciferase Reporter Assay System (Promega) protocol. Luminescence was read using the GloMax Multi Jr Tube Multimode Reader (Promega) with the Luminescence module using the DLR-0INJ protocol. Reporter firefly luciferase luminescence was normalized to renilla luciferase luminescence to generate relative luminescence units (RLUs), calculated by the luminometer, to control for transfection efficiency.
Statistics
For qPCR, quantification and statistical analyses were performed in the BioRad CFX manager 3.0 software. Graphs were generated in Excel 2016 (Microsoft). Sample variance was assessed in Excel 2016 (Microsoft) using Levene’s test and two sample t-tests were performed for pairwise comparisons. Differences in frequencies for immunofluorescence were calculated using χ2 tests.
Results
NPAS3 behaves as predicted for a Class 1 bHLH–PAS transcription factor
As NPAS3 is predicted to be bHLH–PAS transcription factor based on sequence conservation, notably to its orthologue trachealess and its closest paralogue Neuronal PAS-domain containing 1 (Npas1) [2], we set out to determine whether NPAS3 behaves as predicted for a Class 1 bHLH–PAS transcription factor. Class 1 bHLH–PAS proteins obligately interact with Class 2 bHLH–PAS proteins, such as ARNT, through their bHLH and PAS domains [19]. We assessed two isoforms of each NPAS3 and ARNT using the HaloTag system in HEK 293T cells to determine whether they interact with high affinity. Both isoforms of NPAS3 and ARNT were found to interact with one another (Fig. 1b, c). Isoform 1 and 2 of NPAS3, which differ by 30 aa and 2 aa flanking the bHLH DNA binding domain, were both found to interact with ARNT (Fig. 1a, b). Similarly, isoforms 1 and 6 of ARNT, which vary by two residues C-terminal to the bHLH and PAS domains, were also found to be able to interact with both NPAS3 isoforms (Fig. 1b, c). Further, expressed HaloTag-NPAS3 was able to pull-down endogenous ARNT, demonstrating specificity of this interaction (Fig. 1b, d). These data confirm that human NPAS3 and ARNT can interact, as observed for mouse Npas3 and Arnt [22, 23].
In order to experimentally characterize the predicted functional domains of NPAS3, constructs expressing combinations of the bHLH, PAS and/or C-terminal putative transactivation domain were cloned from the NPAS3 isoform 1 coding sequence (Fig. 1a). The interaction with ARNT was found to require both the bHLH and PAS domains, but not the C-terminus (Fig. 2). Immunofluorescence was performed to determine whether domains are localized such that they are able to interact with ARNT. Full length NPAS3 was found to be cytoplasmic and nuclear localized, which was affected by co-expression of ARNT, where nuclear localization was enhanced with ARNT expression (Fig. 3, Additional file 3: Figure S2). All NPAS3 domain constructs were observed in the nucleus, with variable cytoplasmic localization (Fig. 3b). Constructs containing the C-terminus, where a nuclear localization sequence (NLS) has been predicted [13], were found to be predominantly localized to the nucleus in all conditions. Constructs containing the PAS domain were found to have enhanced nuclear localization with co-expression of ARNT. The bHLH domain demonstrated the opposite effect, which may be driven by a nuclear export sequence (NES) predicted in the bHLH domain by NetNES (Fig. 3b, c) [34]. These data suggest that NPAS3 localization may be actively regulated at multiple levels, which can affect gene regulatory output. In summary, our data experimentally validate the function of the predicted domains of NPAS3, and suggest that the observed variable localization of NPAS3 may be contributed to active regulation of NPAS3 localization.

The bHLH and PAS domains are required for high-affinity interaction of NPAS3 and ARNT. Domain constructs used indicated in Fig. 1a. Western blots of HaloTag pull-down of HA-tagged NPAS3 domains by HaloTag-ARNT. Blots include lanes for input sample, unbound sample taken after binding reaction, and protein eluted off of resin after washing. Bands in the top panel of pull-down lane indicate physical association of HA-tagged NPAS3 domain construct with the HaloTag-ARNT construct. Middle and bottom panels are results for the HaloTag-ARNT and HaloTag-empty constructs. As HaloTag constructs are covalently linked to the resin, they will be depleted in the pull-down sample, demonstrating functionality of the HaloTag. Molecular weight of HaloTag-ARNT, 121 kDa, HaloTag construct with no cDNA: 34 kDa. Blot legend with expected sizes: a bHLH–PAS, 49.4 kDa, b PAS-TAD, 98.2 kDa, c bHLH, 13 kDa, d PAS, 35.8 kDa, e TAD, 53.2 kDa. TAD = C-terminal transactivation domain. Pull downs have been replicated at least twice

NPAS3 localization is affected by the region encoding the NLS and independently by ARNT. a Immunofluorescence images demonstrating that NPAS3 is predominantly localized to the nucleus, which is enhanced by co-expression of ARNT. Arrows indicate cytoplasmic localization of NPAS3. ×400 magnification, scale bar = 10 μm. b Quantification of localization of NPAS3 signal. Localization was scored as either nuclear or nuclear = cytoplasmic by an individual blinded to NPAS3 construct identity and to the presence or absence of ARNT. *p < 0.05, **p < 0.01, ***p < 0.001. c Output of NetNES 1.0 prediction software indicating a predicted NES in the region encoding the bHLH domain of NPAS3, indicated by scores above the threshold (red line)
NPAS3 and ARNT co-regulate TXNIP and VGF
As we were able to confirm the interaction between human NPAS3 and ARNT, we screened regulatory targets of NPAS3 for regions likely bound by ARNT to identify potential regulatory targets of the NPAS3::ARNT heterodimer. We screened all genes previously identified as differentially expressed in response to ectopic expression of NPAS3 in human cells for ARNT ENCODE ChIP-seq positive regions within 1 kb of the transcription start site [25, 28]. Thirteen genes were identified in our screen for potential co-targets of NPAS3 and ARNT, listed in Table 1. VGF was added as a potential target due to further characterization in other studies [25, 26]. Of these genes, two were found to be regulated by co-expression of NPAS3 and ARNT: VGF and TXNIP (Fig. 4). The direction of regulation of both VGF (up-regulated) and TXNIP (down-regulated) were found to be consistent with those observed in the index study identifying the regulatory effect [25], however, we did not observe regulation of these targets with singly expressed NPAS3, which was performed in the index study. TXNIP was found to be differentially regulated only in the relatively ‘stressed’ condition where media was not replaced 24 h post transfection, suggesting that this regulation may be stimulus specific. VGF was found to be up-regulated by co-expression of NPAS3 and ARNT in all conditions tested.

NPAS3 and ARNT regulate expression of TXNIP and VGF. a Relative quantity of mRNA as assessed by qRT-PCR in HEK 293T cells expressing NPAS3 or ARNT. Expression was assessed 48 h post-transfection, with media replacement 24 h post-transfection and without. VGF and TXNIP were found to be regulated in response to expression of NPAS3 and ARNT. *p < 0.05, **p < 0.01, ***p < 0.001. Data are representative of three biological replicates. b Western blot demonstrating expression of constructs indicated
NPAS3 and ARNT physically associate with and regulate VGF through its promoter
In order to determine if the observed regulation of VGF is due to association of the NPAS3 with the promoter, HaloCHIP, a variant of ChIP, was performed using HaloTag constructs of NPAS3 and ARNT. NPAS3 was found to bind regions proximal to the promoter of VGF (Fig. 5a, e). A second signal was observed in the distal region probed, which may represent an independent binding event as some signal is observed in the ENCODE ARNT ChIP-seq data (Fig. 5e). ARNT was found to associate with all regions probed at the VGF locus (Fig. 5f). To determine whether the observed binding to the interval upstream of the transcription start site results in the observed upregulation of VGF expression, transactivation assays were performed. Luciferase reporter expression was driven by a construct of the proximal promoter region of VGF. NPAS3 and ARNT were both found to activate reporter expression independently, with additive effects observed with co-expression of isoforms 1 of ARNT and NPAS3 (Fig. 5b). These data indicate that NPAS3 and ARNT physically associate with the VGF proximal promoter resulting in activation of expression.

NPAS3 physically regulates the VGF promoter. a UCSC genome browser data used to identify potential NPAS3::ARNT binding sites proximal to the VGF locus. Accessed from http://genome.ucsc.edu. Top: ConTra v3.0 data indicating the location of predicted ARNT binding sites based on position weight matrices accessed from the JASPAR database. Middle: ENCODE ARNT ChIP-seq data indicating peaks proximal to the VGF locus. Bottom: relative localization of ChIP primers and promoter constructs used to assess regulation of this locus by NPAS3 and ARNT. VGF primers indicate regions amplified corresponding to Distal element (black line), Intron 1 (red line) and Promoter region (green line). b Luciferase data demonstrating that NPAS3 and ARNT regulate expression of the luciferase (luc2) reporter driven by a construct including 797 bp proximal to the transcription start site of VGF. RLU = relative luciferase units, the ratio of firefly luciferase and renilla luciferase to normalize for transfection efficiency. Top and bottom of each box indicate the 25th and 75th centile, internal bar indicates the median, whiskers indicate data extremes. Data are representative of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001. c, d Western blots demonstrating expression of NPAS3 and ARNT constructs. e HaloCHIP PCR data demonstrating that NPAS3 interacts with the promoter region of VGF relative to other regions of the locus assayed. f HaloCHIP PCR data demonstrating ARNT signal at all regions tested at the VGF locus. HaloCHIP results are representative of three replicates
As the VGF promoter construct is regulated by NPAS3 and ARNT, we used it to assess the gene-regulatory activity of the domains of NPAS3 (Fig. 6a). Singly-expressed NPAS3 constructs were not able to activate expression from this locus when the C-terminal predicted transactivation domain was deleted. When co-expressed with ARNT, the NPAS3 bHLH–PAS construct repressed the ability of ARNT to enhance expression of this reporter, supporting the ability of the bHLH–PAS domains to functionally interact with ARNT, and the C-terminus as required for transactivation function. The bHLH domain in isolation also appeared able to repress activation of this reporter by ARNT, potentially suggestive of some interactivity, or ability to compete with ARNT for binding to DNA targets. Assessment of transactivation from a control promoterless luciferase “empty vector” construct demonstrated that the activation observed by the full-length NPAS3 isoform 1 is specific to the VGF promoter region, and that the observed activation in the PAS-TAD and TAD (C-terminus, transactivation domain) constructs is likely due to potent but non-specific transactivation function (Fig. 6b). These data support the role of the C-terminus as a true transactivation domain, as well as of the bHLH domain for conferring sequence specificity of NPAS3 regulatory function.

The contribution of NPAS3 domains to gene regulatory function. a, b Luciferase reporter assay demonstrating the gene-regulatory effects of NPAS3 domains. The C-terminus of NPAS3 is required to transactivate expression of a reporter construct, and without the bHLH and PAS domains the C-terminus drives non-specific transactivation. Reporter used is luciferase (luc2) expression driven by a construct including 797 bp proximal to the transcription start site of VGF. RLU = relative luciferase units, the ratio of firefly luciferase and renilla luciferase to normalize for transfection efficiency. Top and bottom of each box indicate the 25th and 75th centile, internal bar indicates the median, whiskers indicate data extremes. Data are representative of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001. c Expression of NPAS3 domain constructs. Expected sizes: full length: 101 kDa, bHLH–PAS 49.4 kDa, PAS 35.8 kDa, PAS-TAD 98.2 kDa, TAD 53.2 kDa
NPAS3 and ARNT physically associate with and regulate TXNIP
We probed the NPAS3 and ARNT HaloCHIP samples to determine if the observed regulation of TXNIP is associated with physical association of NPAS3 and ARNT. NPAS3 was found to bind regions proximal to the promoter of TXNIP (Fig. 7a, b). Co-expression of ARNT resulted in a secondary signal observed in exon 1, and ARNT was not found to bind specifically associated with the promoter region without expression of NPAS3 (Fig. 7b). Luciferase reporter expression was driven by a TXNIP promoter construct in order to confirm that the observed regulation is driven by binding of NPAS3 this region. The TXNIP promoter could not be generated without secondary variants due to a repetitive C-rich region in the construct. These variants, relative to the transcription start site (Chr1:145,438,461) include − 246hetA/C (Chr1:145,438,215hetA/C), − 222ΔC (Chr1:145,438,239ΔC) and + 49T>C (Chr1:145,438,510T>C) and are indicated in Fig. 7a. These variants were found to lie at regions of low conservation, not predicted to affect ARNT binding, nor within regions characterized as involved in regulation of TXNIP in response to various environmental stimuli such as glucose and oxidative stress [35,36,37,38,39]. As such experiments were performed in low- and high-glucose conditions to confirm responsiveness of the construct to glucose. Co-expression of NPAS3 and ARNT were found to activate expression driven by this construct (Fig. 7c, d). The promoter was found to be responsive to glucose concentration, with higher baseline expression in high glucose relative to low glucose. The up-regulation driven by NPAS3 and ARNT was observed in both low- and high-glucose conditions. These data are opposite to the repression of the endogenous TXNIP locus by NPAS3 and ARNT co-expression observed by us, and in the index study identifying TXNIP as down-regulated by NPAS3 (Fig. 4, [25]). However, these data support physical association of NPAS3 and ARNT to the region encoded in this construct, despite the region cloned not being sufficient to assemble the repressive complex.

NPAS3 and ARNT physically regulate the TXNIP locus. a UCSC genome browser data used to identify potential NPAS3::ARNT binding sites proximal to the TXNIP locus. Accessed from http://genome.ucsc.edu. Top: ConTra v3.0 predicted ARNT binding sites in the promoter, 3′UTR and intron 1 of TXNIP based on position weight matrices accessed from the JASPAR database. Middle: ENCODE ARNT ChIP-seq data indicating peaks proximal to the TXNIP locus. Bottom: relative localization of ChIP primers and promoter constructs used to assess regulation of this locus by NPAS3 and ARNT. TXNIP primers indicate regions amplified corresponding to Distal element (black line), Exon 1 (red line) and Promoter region (green line). Red asterisks indicate variants in promoter construct relative to wild-type sequence. b Luciferase data demonstrating that NPAS3 and ARNT regulate expression of reporter luciferase (luc2) driven by a construct including 867 bp proximal to the transcription start site of TXNIP. RLU = relative luciferase units, the ratio of firefly luciferase and renilla luciferase to normalize for transfection efficiency. Top and bottom of each box indicate the 25th and 75th centile, internal bar indicates the median. Data are presented for two biological replicates performed in each low- and high-glucose conditions. *p < 0.05, **p < 0.01, ***p < 0.001. c, d Western blots demonstrating expression of NPAS3 and ARNT constructs. e HaloCHIP PCR data demonstrating that NPAS3 interacts with the promoter region of VGF relative to other regions of the locus assayed. f HaloCHIP PCR data demonstrating ARNT signal at all regions tested at the VGF locus. HaloCHIP results are representative of three replicates performed
Functional assessment of NPAS3 variants
Multiple coding variants of NPAS3 have been identified in human studies, including rare and common variants associated with psychiatric illness (p.Val304Ile [12] and p.Ala552Pro [13], respectively). We functionally assessed these variants using the developed assays, as well as a rare population variant, p.Gly697Ser [13]. The rare psychiatric illness-associated p.Val304Ile is localized between the PAS domains of NPAS3 and has been shown to be associated with aggregation, further, it can be hypothesized to affect protein interactivity and function (Fig. 1a, [40]). Although our assays were not designed to assess protein aggregation, we did not observe alterations in expression, localization, deficits in interactivity with ARNT or transactivation function due to this variant in our assays (Figs. 8, 9a–c). For the schizophrenia-associated variant p.Ala552Pro, localized to the transactivation domain, we did not observe functional consequences in any of our assays (Figs. 8, 9a, b, d). For the rare variant localized to a poly-glycine repeat in the transactivation domain not associated with disorder, p.Gly697Ser, we observed normal expression, localization and transactivation function when expressed in isolation, however, co-expression with ARNT resulted in reduced transactivation output (Figs. 8, 9a, b, d).

Localization of NPAS3 variants. Immunofluorescence microscopy images demonstrating that all NPAS3 variants are able to localize to the nucleus. HEK 293T cells were transfected with indicated constructs and incubated for 48 h prior to fixing with paraformaldehyde and probing for ARNT and NPAS3, and stained with DAPI to indicate nuclei. ×400 magnification, scale bar = 10 μm

Functional characterization of NPAS3 variants. a Western blots showing that NPAS3 variants are normally expressed when transfected into HEK 293T cells. b HaloTag pull-down data demonstrating that the p.Val304Ile and p.Ala552Pro variants are able to interact with ARNT. Bands in the top panel of pull-down lane of ‘prey’ protein, indicating physical association of HA-NPAS3 variants with the HaloTag-ARNT construct. Middle and bottom panels are results for the HaloTag-clone and HaloTag-empty constructs. As HaloTag constructs are covalently linked to the resin, they will be depleted in the pull-down sample, demonstrating functionality of the HaloTag. Molecular weight of HaloTag-ARNT, 121 kDa, HaloTag construct with no cDNA: 34 kDa, Full-length NPAS3: 101 kDa. c, d Luciferase reporter expression demonstrating transactivation function of all three variants tested. The previously characterized variant p.Val304Ile was tested in two isoforms in (c). The remaining variants, p.Ala552Pro and p.Gly697Ser was tested in isoform 1. Reporter used is luciferase (luc2) expression driven by a construct including 797 bp proximal to the transcription start site of VGF. RLU = relative luciferase units, the ratio of firefly luciferase and renilla luciferase to normalize for transfection efficiency. Top and bottom of each box indicate the 25th and 75th centile, internal bar indicates the median, whiskers indicate data extremes. Data are representative of three biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Through these studies we have experimentally demonstrated that NPAS3 acts as a true transcription factor, behaving as predicted for a bHLH–PAS family member. NPAS3 has been characterized as a bHLH–PAS transcription factor requiring ARNT as an obligate heterodimeric partner, however, the interaction between NPAS3 and ARNT had not been experimentally demonstrated until recent studies of murine Npas3 and Arnt [22, 23]. Our data confirm that the interaction is conserved for human NPAS3 and ARNT, and that it requires both the bHLH and PAS domains. We observe similar effects of ARNT expression on NPAS3 expression as observed for NPAS1, except NPAS3 is much more strongly associated with the nucleus than NPAS1 [49]. As NPAS3 has been observed to be variably localized to the nucleus by immunohistochemistry and immunofluorescence, this may indicate that subcellular localization of NPAS3 is an active process that is regulated in response to environmental or cellular stimuli [12, 14, 25, 50]. Interestingly, we have observed an effect of ARNT expression on the localization of the NPAS3 PAS domain construct in isolation, which we did not observe to be able to interact with ARNT in our pull-down studies. This may be due to the detection of a weak residual interactivity in in cellulo, which is not of high enough affinity to be captured by our pull down assay. Alternately, expression of ARNT may enhance nuclear localization of the PAS domain of NPAS3 by interacting with and saturating the pool of a cytoplasmic protein involved in the regulation of bHLH–PAS protein localization and preventing it from restricting the NLS-deficient PAS construct to the cytoplasm.
We have experimentally validated the categorization of NPAS3 as a transcription factor through assessment of its ability to bind DNA and cause regulation of target genes. To this end, we co-registered ARNT ChIP-seq data with previously generated microarray data screening for genes differentially regulated by NPAS3 [25, 30]. Of the 14 genes identified as potential co-targets, two were found to be regulated by expression of NAPS3 and ARNT: TXNIP and VGF. The regulation of VGF as a target of NPAS3 has been assessed previously, however, the contribution of ARNT to this regulation has not been studied [25, 26, 51]. Similar to other groups, we observe that expression of NPAS3 results in activation of reporter expression driven by VGF promoter constructs. Our construct is limited to the 797 bp of the proximal VGF promoter, excluding most of exon 1 and intron 1 entirely, and expression of NPAS3 results in the same magnitude of activation as the larger constructs used by other groups. Combined with our ChIP data, we have shown that NPAS3 associates with the proximal promoter region to regulate VGF expression. We further find that co-expression of NPAS3 and ARNT results in additive effects on VGF promoter driven expression.
Repression of TXNIP by NPAS3 was replicated in our studies but only with co-expression of NPAS3 and ARNT [25]. In other experiments, we have observed that TXNIP mRNA expression increases over time in culture (data not shown). As TXNIP is rapidly regulated to maintain metabolic homeostasis and apoptotic response to multiple environmental stresses and cues, this may represent a cellular response to altered metabolic state and increased propensity to apoptosis in the face of environmental stress due to media depletion, pH changes, reduced levels of nutrients, increases in metabolic by-products and oxidative stress over time in culture [52,53,54,55,56]. NPAS3 may inhibit the induction of TXNIP in order to promote cellular survival, similar to its pro-neurogenic role in the hippocampus [15]. NPAS3 was found to associate with a region proximal to the promoter of TXNIP. TXNIP regulation was found to require co-expression of NPAS3 and ARNT, however, the region including the 867 bp 5′ of the transcription start site was found not to be sufficient for repression of reporter expression. Exploration of other binding sites proximal to this region, including exon 1/intron 1 should be undertaken in order to further characterize the nature of the repressive complex. ARNT may bind at a distal site to facilitate repression, which may explain why we observe binding of NPAS3 to the TXNIP promoter in the absence of expressed ARNT and gene regulatory output. Although our construct contains variants relative to the genomic locus, these variants are outside of predicted ARNT binding sites, and the construct was found to be expressive, and normally responsive to glucose, suggesting that these variants do not significantly affect function [57, 58].
A recent ChIP-seq study of the mouse hippocampus, where deletion of Npas3 has been associated with loss of neurogenesis, did not identify VGF or TXNIP as differentially regulated by loss of NPAS3, nor bound by NPAS3 in wild-type mouse hippocampus [27]. This may represent a difference due to differences in methodology and systems assessed, as we have used an in vitro human cell system. As this model system is markedly different in cell type (monoculture of human cells derived from embryonic kidney) and cultured in synthetic medium, the gene regulatory responses elicited by NPAS3 expression may vary markedly, potentially due to environmental factors as well as varying expression of potential gene regulatory co-factors that affect gene regulation by NPAS3. We selected HEK 293T cells as they are directly related to the cells the original study used to identify NPAS3-regulated genes in order to assess the mechanism of regulation by NPAS3 that results in the observed changes in gene expression [25]. Transcriptomic profiling suggests that these cells are of neural crest origin and express neural genes [59]. Further, Npas3 is expressed in non-neural cells and its gene-regulatory mechanism is relevant to multiple tissue- and cell-types [2, 24, 60]. The non-replication may also be contributed to by variable regulation in response to environmental stimuli, as we observe regulation of TXNIP only under stress conditions. TXNIP has been shown to be rapidly and transiently regulated in response to cellular stimuli, as such occupancy of NPAS3 at this locus may not be constitutive [61]. As TXNIP is involved in cellular redox balance, and response of cells to intra- and extracellular stressors, affecting inflammatory tone, metabolism and apoptotic pathways, NPAS3 may contribute to cellular survival by repressing the pro-apoptotic function of TXNIP [54, 55, 62, 63].
Using the assays developed to characterize full-length NPAS3 and its interacting partner, ARNT, we have characterized the predicted functional domains of NPAS3. The bHLH domain was found to be critical for interaction with ARNT, as well as for specificity of the regulatory action of NPAS3. The bHLH domain contains a predicted nuclear export sequence and appears to contribute to the subcellular localization of NPAS3. Both the PAS and the bHLH domains were found to be critical for heterodimerization with ARNT, consistent with the observed interaction interfaces identified in the crystal structure of Npas3::Arnt and other bHLH–PAS heterodimers [23, 64, 65]. The PAS(A) domain is considered to be critical for the specific and high-affinity interaction of bHLH–PAS proteins with ARNT [20, 21] while the PAS(B) domains of bHLH–PAS proteins are critical for normal gene regulatory function, ligand binding and protein::protein interactions with chaperone proteins [21, 66,67,68,69]. Both PAS(A) and PAS(B) domains have been shown to be involved in interactions with co-activators of bHLH–PAS proteins [70,71,72]. Although we did not assess the PAS(A) and PAS(B) repeats independently, other groups have assessed a construct encoding the bHLH and PAS(A) domain independently and find that it can effect muted gene regulatory function relative to full-length NPAS3 [25, 26, 51]. The reduced gene regulatory output is likely contributed to by loss of coactivators potentially recruited by the PAS(B) and C-terminal transactivation domain, and potentially due to reduced interactivity with heterodimeric partners, such as ARNT. Our data demonstrate that the region C-terminal to the PAS(B) domain predominantly contributes to the transactivation function of NPAS3. The bHLH–PAS domain construct studied here acts to repress activation by ARNT, and is unable to cause activation of reporter expression when expressed in isolation. Furthermore, expression of the C-terminus in isolation is sufficient to non-specifically activate reporter gene expression, demonstrating potent transactivation function. These data confirm for the first time that the C-terminus of NPAS3 encodes a true transactivation domain. Through these studies we have confirmed the function of the domains of NPAS3, and have experimentally demonstrated that NPAS3 acts as a true transcription factor.
Finally, we assayed three variants identified in the human population for effects on NPAS3 function. The psychiatric disorder-associated variants p.Val304Ile and p.Ala552Pro were found to be normally expressed and localized to the nucleus, and further, did not affect interaction with ARNT, nor regulation of the VGF reporter construct relative to wild-type. Previous studies have identified that the p.Val304Ile variant, which has been found to be sequestered in the insoluble fraction of cell lysates, suggestive of aggregation [51]. This variant has been observed in various populations at low frequency [Exome Aggregation Consortium (ExAC) worldwide minor allele frequency (MAF) = 0.0001] [73]. Our studies were not designed to assess aggregation, however, we found no reduction in its ability to activate expression driven the VGF promoter which is in conflict with previously observed deficits [51]. As such further study of this variant is warranted to validate the effects of this variant on NPAS3 function.
Since its association with schizophrenia, the p.Ala552Pro variant has since been found at similar frequencies in unselected and normal control populations (ExAC worldwide MAF = 0.14) [13, 17, 73]. We undertook studies to functionally characterize this variant due to the nature of the amino acid substitution, a large proline residue in place of an alanine, which have been shown to be poorly tolerated [74]. We did not observe any functional effect of this variant, which may be expected due to its presence in the normal population. Our assays may not be sensitive enough to detect the functional significance of this variant, or may not be designed in such a way as to detect the effects, for example, the predicted alteration to splicing enhancers [13].
Finally we assessed the low frequency variant p.Gly697Ser, which has not been associated with disorder, but is present in the normal population at a low frequency (ExAC worldwide MAF = 0.0013) [13, 73]. This variant was found to be normally expressed and localized to the nucleus. Expressed individually, the transactivation function of this variant was found to be normal, however, when co-expressed with ARNT, it was found not to cooperatively activate expression driven from the VGF promoter. This variant is localized to a poly glycine repeat within the transactivation domain which has been expanded in humans [13, 18]. Although the function of poly-glycine repeats is poorly characterized, it is thought to be involved in spacing of functional domains and may contribute to protein::protein interactions, based on observations of aggregation associated with large expansions [75,76,77]. Variants as small as 1 amino acid deletions in poly-glycine repeats have been shown to affect protein function, and have been associated with human disorders [78,79,80]. As such this variant may contribute to variation in NPAS3 transactivation function, potentially by affecting interaction with a co-activator of the NPAS3::ARNT heterodimer, or other functional NPAS3 complex.
Conclusions
Through these studies we have demonstrated that NPAS3 acts as a transcription factor. Furthermore, we have experimentally validated the function of the bHLH domain in DNA binding, the PAS domains in interaction with ARNT, and C-terminal tail as a potent transactivation domain. In order to expand our understanding of variation to NPAS3, we characterized the functional significance of variants identified in the human population. We found that both the previously psychiatric disorder-associated variants, p.Val304Ile and p.Ala552Pro variants did not have significantly altered molecular function. However, we identified altered transactivation function for the rare population variant p.Gly697Ser when co-expressed with ARNT. These data expand our understanding of the molecular function of NPAS3, as well as the contribution of variants to NPAS3 in its gene regulatory function and are important for interpretation of variants identified in next generation sequencing studies of individuals.
References
Kamnasaran D, Muir WJ, Ferguson-Smith MA, Cox DW. Disruption of the neuronal PAS3 gene in a family affected with schizophrenia. J Med Genet. 2003;40(5):325–32.
Brunskill EW, Witte DP, Shreiner AB, Potter SS. Characterization of npas3, a novel basic helix-loop-helix PAS gene expressed in the developing mouse nervous system. Mech Dev. 1999;88(2):237–41.
Pickard BS, Malloy MP, Porteous DJ, Blackwood DH, Muir WJ. Disruption of a brain transcription factor, NPAS3, is associated with schizophrenia and learning disability. Am J Med Genet B Neuropsychiatr Genet. 2005;136B(1):26–32.
Kamnasaran D, Chen CP, Devriendt K, Mehta L, Cox DW. Defining a holoprosencephaly locus on human chromosome 14q13 and characterization of potential candidate genes. Genomics. 2005;85(5):608–21.
Rosenfeld JA, Ballif BC, Martin DM, Aylsworth AS, Bejjani BA, Torchia BS, Shaffer LG. Clinical characterization of individuals with deletions of genes in holoprosencephaly pathways by aCGH refines the phenotypic spectrum of HPE. Hum Genet. 2010;127(4):421–40.
Piccione M, Serra G, Consiglio V, Di Fiore A, Cavani S, Grasso M, Malacarne M, Pierluigi M, Viaggi C, Corsello G. 14q13.1-21.1 deletion encompassing the HPE8 locus in an adolescent with intellectual disability and bilateral microphthalmia, but without holoprosencephaly. Am J Med Genet A. 2012;158A(6):1427–33.
Phelps R, Tsai A, Hagen A, Pinter J, Smith R, Stein MT. The curse of the dolphins: cognitive decline and psychosis. J Dev Behav Pediatr. 2016;37(4):343–5.
Visser R, Gijsbers A, Ruivenkamp C, Karperien M, Reeser HM, Breuning MH, Kant SG, Wit JM. Genome-wide SNP array analysis in patients with features of sotos syndrome. Horm Res Paediatr. 2010;73(4):265–74.
Gupta CN, Chen J, Liu J, Damaraju E, Wright C, Perrone-Bizzozero NI, Pearlson G, Luo L, Michael AM, Turner JA, Calhoun VD. Genetic markers of white matter integrity in schizophrenia revealed by parallel ICA. Front Hum Neurosci. 2015;9:100.
Weber H, Kittel-Schneider S, Gessner A, Domschke K, Neuner M, Jacob CP, Buttenschon HN, Boreatti-Hummer A, Volkert J, Herterich S, Baune BT, Gross-Lesch S, Kopf J, Kreiker S, Nguyen TT, Weissflog L, Arolt V, Mors O, Deckert J, Lesch KP, Reif A. Cross-disorder analysis of bipolar risk genes: further evidence of DGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology. 2011;36(10):2076–85.
Nurnberger JI Jr, Koller DL, Jung J, Edenberg HJ, Foroud T, Guella I, Vawter MP, Kelsoe JR, Psychiatric Genomics Consortium Bipolar Group. Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry. 2014;71(6):657–64.
Yu L, Arbez N, Nucifora LG, Sell GL, Delisi LE, Ross CA, Margolis RL, Nucifora FC Jr. A mutation in NPAS3 segregates with mental illness in a small family. Mol Psychiatry. 2014;19(1):7–8.
Macintyre G, Alford T, Xiong L, Rouleau GA, Tibbo PG, Cox DW. Association of NPAS3 exonic variation with schizophrenia. Schizophr Res. 2010;120(1–3):143–9.
Erbel-Sieler C, Dudley C, Zhou Y, Wu X, Estill SJ, Han T, Diaz-Arrastia R, Brunskill EW, Potter SS, McKnight SL. Behavioral and regulatory abnormalities in mice deficient in the NPAS1 and NPAS3 transcription factors. Proc Natl Acad Sci USA. 2004;101(37):13648–53.
Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA, Richardson JA, Brat DJ, McKnight SL. The neuronal PAS domain protein 3 transcription factor controls FGF-mediated adult hippocampal neurogenesis in mice. Proc Natl Acad Sci USA. 2005;102(39):14052–7.
Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De Brabander J, Ready JM, McKnight SL. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142(1):39–51.
Stanco A, Pla R, Vogt D, Chen Y, Mandal S, Walker J, Hunt RF, Lindtner S, Erdman CA, Pieper AA, Hamilton SP, Xu D, Baraban SC, Rubenstein JL. NPAS1 represses the generation of specific subtypes of cortical interneurons. Neuron. 2014;84(5):940–53.
Pickard BS, Pieper AA, Porteous DJ, Blackwood DH, Muir WJ. The NPAS3 gene–emerging evidence for a role in psychiatric illness. Ann Med. 2006;38(6):439–48.
Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36(2):189–204.
McIntosh BE, Hogenesch JB, Bradfield CA. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol. 2010;72:625–45.
Pongratz I, Antonsson C, Whitelaw ML, Poellinger L. Role of the PAS domain in regulation of dimerization and DNA binding specificity of the dioxin receptor. Mol Cell Biol. 1998;18(7):4079–88.
Moen MJ, Adams HH, Brandsma JH, Dekkers DH, Akinci U, Karkampouna S, Quevedo M, Kockx CE, Ozgur Z, van IJcken WF, Demmers J, Poot RA. An interaction network of mental disorder proteins in neural stem cells. Transl Psychiatry. 2017;7(4):e1082.
Wu D, Su X, Potluri N, Kim Y, Rastinejad F. NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH–PAS family as multi-ligand binding transcription factors. Elife. 2016. https://doi.org/10.7554/elife.18790.
Zhou S, Degan S, Potts EN, Foster WM, Sunday ME. NPAS3 is a trachealess homolog critical for lung development and homeostasis. Proc Natl Acad Sci USA. 2009;106(28):11691–6.
Sha L, Macintyre L, Machell JA, Kelly MP, Porteous DJ, Brandon NJ, Muir WJ, Blackwood DH, Watson DG, Clapcote SJ, Pickard BS. Transcriptional regulation of neurodevelopmental and metabolic pathways by NPAS3. Mol Psychiatry. 2012;17(3):267–79.
Yang D, Zhang W, Padhiar A, Yue Y, Shi Y, Zheng T, Davis K, Zhang Y, Huang M, Li Y, Sha L. NPAS3 regulates transcription and expression of VGF: implications for neurogenesis and psychiatric disorders. Front Mol Neurosci. 2016;9:109.
Michaelson JJ, Shin MK, Koh JY, Brueggeman L, Zhang A, Katzman A, McDaniel L, Fang M, Pufall M, Pieper AA. Neuronal PAS domain proteins 1 and 3 are master regulators of neuropsychiatric risk genes. Biol Psychiatry. 2017. https://doi.org/10.1016/j.biopsych.2017.03.021.
Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J, Throop A, Fiacco M, LaBaer J. DNASU plasmid and PSI: biology-materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42(Database issue):D1253–60.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, Lee BT, Barber GP, Harte RA, Diekhans M, Long JC, Wilder SP, Zweig AS, Karolchik D, Kuhn RM, Haussler D, Kent WJ. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41(Database issue):D56–63.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006.
Pchelintsev NA, Adams PD, Nelson DM. Critical parameters for efficient sonication and improved chromatin immunoprecipitation of high molecular weight proteins. PLoS ONE. 2016;11(1):e0148023.
Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, Hickey G, Hinrichs AS, Hubley R, Karolchik D, Learned K, Lee BT, Li CH, Miga KH, Nguyen N, Paten B, Raney BJ, Smit AF, Speir ML, Zweig AS, Haussler D, Kuhn RM, Kent WJ. The UCSC genome browser database: 2015 update. Nucleic Acids Res. 2015;43(Database issue):D670–81.
Kreft L, Soete A, Hulpiau P, Botzki A, Saeys Y, De Bleser P. ConTra v3: a tool to identify transcription factor binding sites across species, update 2017. Nucleic Acids Res. 2017. https://doi.org/10.1093/nar/gkx376.
Zhang W, Ni C, Sheng J, Hua Y, Ma J, Wang L, Zhao Y, Xing Y. TLQP-21 protects human umbilical vein endothelial cells against high-glucose-induced apoptosis by increasing G6PD expression. PLoS ONE. 2013;8(11):e79760.
Severini C, Ciotti MT, Biondini L, Quaresima S, Rinaldi AM, Levi A, Frank C, Possenti R. TLQP-21, a neuroendocrine VGF-derived peptide, prevents cerebellar granule cells death induced by serum and potassium deprivation. J Neurochem. 2008;104(2):534–44.
Saade E, Mechold U, Kulyyassov A, Vertut D, Lipinski M, Ogryzko V. Analysis of interaction partners of H4 histone by a new proteomics approach. Proteomics. 2009;9(21):4934–43.
Ishihara S, Yasuda M, Ishizu A, Ishikawa M, Shirato H, Haga H. Activating transcription factor 5 enhances radioresistance and malignancy in cancer cells. Oncotarget. 2015;6(7):4602–14.
Torres-Peraza JF, Engel T, Martin-Ibanez R, Sanz-Rodriguez A, Fernandez-Fernandez MR, Esgleas M, Canals JM, Henshall DC, Lucas JJ. Protective neuronal induction of ATF5 in endoplasmic reticulum stress induced by status epilepticus. Brain. 2013;136(Pt 4):1161–76.
Hernandez-Jimenez M, Martinez-Lopez D, Gabande-Rodriguez E, Martin-Segura A, Lizasoain I, Ledesma MD, Dotti CG, Moro MA. Seladin-1/DHCR24 is neuroprotective by associating EAAT2 glutamate transporter to lipid rafts in experimental stroke. Stroke. 2016;47(1):206–13.
Haffner C, Dettmer U, Weiler T, Haass C. The Nicastrin-like protein Nicalin regulates assembly and stability of the Nicalin-nodal modulator (NOMO) membrane protein complex. J Biol Chem. 2007;282(14):10632–8.
Liu Q, Liu L, Zhao Y, Zhang J, Wang D, Chen J, He Y, Wu J, Zhang Z, Liu Z. Hypoxia induces genomic DNA demethylation through the activation of HIF-1alpha and transcriptional upregulation of MAT2A in hepatoma cells. Mol Cancer Ther. 2011;10(6):1113–23.
Zhang Z, Jones A, Joo HY, Zhou D, Cao Y, Chen S, Erdjument-Bromage H, Renfrow M, He H, Tempst P, Townes TM, Giles KE, Ma L, Wang H. USP49 deubiquitinates histone H2B and regulates cotranscriptional pre-mRNA splicing. Genes Dev. 2013;27(14):1581–95.
Li S, Sheng J, Hu JK, Yu W, Kishikawa H, Hu MG, Shima K, Wu D, Xu Z, Xin W, Sims KB, Landers JE, Brown RH Jr, Hu GF. Ribonuclease 4 protects neuron degeneration by promoting angiogenesis, neurogenesis, and neuronal survival under stress. Angiogenesis. 2013;16(2):387–404.
Daftuar L, Zhu Y, Jacq X, Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS ONE. 2013;8(7):e68667.
Yoshihara E, Masaki S, Matsuo Y, Chen Z, Tian H, Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514.
Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37(14):4587–602.
Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, Espinosa JM. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153(6):1327–39.
Teh CH, Lam KK, Loh CC, Loo JM, Yan T, Lim TM. Neuronal PAS domain protein 1 is a transcriptional repressor and requires arylhydrocarbon nuclear translocator for its nuclear localization. J Biol Chem. 2006;281(45):34617–29.
Gould P, Kamnasaran D. Immunohistochemical analyses of NPAS3 expression in the developing human fetal brain. Anat Histol Embryol. 2011. https://doi.org/10.1111/j.1439-0264.2010.01059.x.
Nucifora LG, Wu YC, Lee BJ, Sha L, Margolis RL, Ross CA, Sawa A, Nucifora FC Jr. A mutation in NPAS3 that segregates with schizophrenia in a small family leads to protein aggregation. Mol Neuropsychiatry. 2016;2(3):133–44.
Kim GS, Jung JE, Narasimhan P, Sakata H, Chan PH. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiol Dis. 2012;46(2):440–9.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11(2):136–40.
Hui ST, Andres AM, Miller AK, Spann NJ, Potter DW, Post NM, Chen AZ, Sachithanantham S, Jung DY, Kim JK, Davis RA. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc Natl Acad Sci USA. 2008;105(10):3921–6.
Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol. 2000;164(12):6287–95.
Wang Y, De Keulenaer GW, Lee RT. Vitamin D(3)-up-regulated protein-1 is a stress-responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem. 2002;277(29):26496–500.
Cha-Molstad H, Saxena G, Chen J, Shalev A. Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem. 2009;284(25):16898–905.
Yu FX, Chai TF, He H, Hagen T, Luo Y. Thioredoxin-interacting protein (Txnip) gene expression: sensing oxidative phosphorylation status and glycolytic rate. J Biol Chem. 2010;285(33):25822–30.
Lin YC, Boone M, Meuris L, Lemmens I, Van Roy N, Soete A, Reumers J, Moisse M, Plaisance S, Drmanac R, Chen J, Speleman F, Lambrechts D, Van de Peer Y, Tavernier J, Callewaert N. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat Commun. 2014;5:4767.
Kamm GB, Pisciottano F, Kliger R, Franchini LF. The developmental brain gene NPAS3 contains the largest number of accelerated regulatory sequences in the human genome. Mol Biol Evol. 2013;30(5):1088–102.
Chai TF, Leck YC, He H, Yu FX, Luo Y, Hagen T. Hypoxia-inducible factor independent down-regulation of thioredoxin-interacting protein in hypoxia. FEBS Lett. 2011;585(3):492–8.
Abderrazak A, Syrovets T, Couchie D, El Hadri K, Friguet B, Simmet T, Rouis M. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307.
Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, Greiner D, Kaufman RJ, Bortell R, Urano F. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16(2):265–73.
Seok SH, Lee W, Jiang L, Molugu K, Zheng A, Li Y, Park S, Bradfield CA, Xing Y. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc Natl Acad Sci USA. 2017;114(21):5431–6.
Wu D, Potluri N, Lu J, Kim Y, Rastinejad F. Structural integration in hypoxia-inducible factors. Nature. 2015;524(7565):303–8.
McGuire J, Okamoto K, Whitelaw ML, Tanaka H, Poellinger L. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: delineation of overlapping repression and ligand binding functions within the PAS domain. J Biol Chem. 2001;276(45):41841–9.
McGuire J, Whitelaw ML, Pongratz I, Gustafsson JA, Poellinger L. A cellular factor stimulates ligand-dependent release of hsp90 from the basic helix-loop-helix dioxin receptor. Mol Cell Biol. 1994;14(4):2438–46.
Lindebro MC, Poellinger L, Whitelaw ML. Protein-protein interaction via PAS domains: role of the PAS domain in positive and negative regulation of the bHLH/PAS dioxin receptor-Arnt transcription factor complex. EMBO J. 1995;14(14):3528–39.
Hayasaka K, Kitanishi K, Igarashi J, Shimizu T. Heme-binding characteristics of the isolated PAS-B domain of mouse Per2, a transcriptional regulatory factor associated with circadian rhythms. Biochim Biophys Acta. 2011;1814(2):326–33.
Sullivan AE, Peet DJ, Whitelaw ML. MAGED1 is a novel regulator of a select subset of bHLH PAS transcription factors. FEBS J. 2016;283(18):3488–502.
Guo Y, Scheuermann TH, Partch CL, Tomchick DR, Gardner KH. Coiled-coil coactivators play a structural role mediating interactions in hypoxia-inducible factor heterodimerization. J Biol Chem. 2015;290(12):7707–21.
Partch CL, Gardner KH. Coactivators necessary for transcriptional output of the hypoxia inducible factor, HIF, are directly recruited by ARNT PAS-B. Proc Natl Acad Sci USA. 2011;108(19):7739–44.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Gray VE, Hause RJ, Fowler DM. Analysis of large-scale mutagenesis data to assess the impact of single amino acid substitutions. Genetics. 2017. https://doi.org/10.1534/genetics.117.300064.
Alba MM, Guigo R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 2004;14(4):549–54.
Mojsin M, Kovacevic-Grujicic N, Krstic A, Popovic J, Milivojevic M, Stevanovic M. Comparative analysis of SOX3 protein orthologs: expansion of homopolymeric amino acid tracts during vertebrate evolution. Biochem Genet. 2010;48(7–8):612–23.
Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78(3):440–55.
Brito M, Malta-Vacas J, Carmona B, Aires C, Costa P, Martins AP, Ramos S, Conde AR, Monteiro C. Polyglycine expansions in eRF3/GSPT1 are associated with gastric cancer susceptibility. Carcinogenesis. 2005;26(12):2046–9.
Brockschmidt FF, Nothen MM, Hillmer AM. The two most common alleles of the coding GGN repeat in the androgen receptor gene cause differences in protein function. J Mol Endocrinol. 2007;39(1):1–8.
Harvey CG, Menon SD, Stachowiak B, Noor A, Proctor A, Mensah AK, Mnatzakanian GN, Alfred SE, Guo R, Scherer SW, Kennedy JL, Roberts W, Srivastava AK, Minassian BA, Vincent JB. Sequence variants within exon 1 of MECP2 occur in females with mental retardation. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(3):355–60.
Authors’ contributions
LML wrote the manuscript, designed, performed analyzed all experiments. FBB provided supervision, contributed to experimental design and revised the manuscript. Both authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge the input from Georgina Macintyre, including a gift of the HaloTag-ARNT construct.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its Additional information files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This project was funded through Canadian Institutes for Health Research grants number MOP114921 and MOP200810. FBB holds the Shriners Hospital for Children Endowed Chair in Pediatric Scoliosis Research. LML was funded through studentships from Alberta Innovates (formerly Alberta Heritage Fund for Medical Research), the Canadian Institutes for Health Research and the National Science and Engineering Research Council.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Validation of NPAS3 antibody. (A) Western blot demonstrating detection of both NPAS3 constructs used in this study, with co-detection by the HA antibody. (B) Western blot demonstrating detection of expressed NPAS3, as well as a 100 kDa band in SK-N-SH cells known to express NPAS3, no NPAS3 antibody bands are co-detected by the NPAS1 antibody. (C) Immunoprecpititation data demonstrating that the NPAS3 antibody can immunoprecipitate expressed NPAS3 constructs. (D) Immunofluorescence with the NPAS3 antibody detected predominantly nuclear signal which did not overlap with NPAS1 signal, which was predominantly cytoplasmic. 1000X magnification, scale bar = 5 μm.
Additional file 2: Table S1.
Primers used for qPCR analysis of gene expression in this study. Table S2. Primers used for ChIP PCR analysis.
Additional file 3: Figure S2.
Immunofluorescence images of NPAS3 domains. Immunofluorescence microscopy images of cells expressing NPAS3 domain constructs in the presence and absence of co-expressed ARNT. HEK 293T cells were transfected with indicated constructs and incubated for 48 hours prior to fixing with paraformaldehyde and probing for ARNT and HA (NPAS3 domain constructs), and stained with DAPI to indicate nuclei. 400X magnification, scale bar = 10 μm.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Luoma, L.M., Berry, F.B. Molecular analysis of NPAS3 functional domains and variants. BMC Molecular Biol 19, 14 (2018). https://doi.org/10.1186/s12867-018-0117-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12867-018-0117-4