
Abstract
Background
Recurrent aphthous stomatitis (RAS) is a common oral mucosal disorder of unclear etiopathogenesis. Although recent studies of the oral microbiota by high-throughput sequencing of 16S rRNA genes have suggested that imbalances in the oral microbiota may contribute to the etiopathogenesis of RAS, no specific bacterial species associated with RAS have been identified. The present study aimed to characterize the microbiota in the oral mucosa and saliva of RAS patients in comparison with control subjects at the species level.
Results
The bacterial communities of the oral mucosa and saliva from RAS patients with active lesions (RAS, n = 18 for mucosa and n = 8 for saliva) and control subjects (n = 18 for mucosa and n = 7 for saliva) were analyzed by pyrosequencing of the 16S rRNA genes. There were no significant differences in the alpha diversity between the controls and the RAS, but the mucosal microbiota of the RAS patients showed increased inter-subject variability. A comparison of the relative abundance of each taxon revealed decreases in the members of healthy core microbiota but increases of rare species in the mucosal and salivary microbiota of RAS patients. Particularly, decreased Streptococcus salivarius and increased Acinetobacter johnsonii in the mucosa were associated with RAS risk. A dysbiosis index, which was developed using the relative abundance of A. johnsonii and S. salivarius and the regression coefficients, correctly predicted 83 % of the total cases for the absence or presence of RAS. Interestingly, A. johnsonii substantially inhibited the proliferation of gingival epithelial cells and showed greater cytotoxicity against the gingival epithelial cells than S. salivarius.
Conclusion
RAS is associated with dysbiosis of the mucosal and salivary microbiota, and two species associated with RAS have been identified. This knowledge may provide a diagnostic tool and new targets for therapeutics for RAS.
Similar content being viewed by others


Background
Recurrent aphthous stomatitis (RAS) is one of the most common oral mucosal disorders affecting at least 10 to 20 % of the general population [1]. RAS is characterized by the recurrent occurrence of well-circumscribed, single or multiple ulcers that are extremely painful and heal more slowly than traumatic ulcers of similar size [2]. Diverse factors, including genetic predisposition, immunologic disturbances, viral and bacterial infections, food allergies, vitamin and microelement deficiencies, systemic diseases, hormonal imbalance, mechanical injuries, and stress, have been suggested to trigger or to be associated with RAS [3]. However, the etiopathogenesis of RAS remains unclear. Consequently, no curative treatment is available and patient care primarily consists of symptomatic treatment [1].
Among the bacterial infections, the role of a Streptococcus strain (first identified as S. sanguinis but now reclassified as S. oralis) has been extensively studied since its isolation from a RAS lesion. Cross-reaction of anti-Streptococcal antibodies with autoantigens in the oral mucosa was proposed as an etiopathogenic mechanism of RAS, but no evidence to support this hypothesis has been found [4]. Later, lack of association between S. oralis and RAS was reported based on the less frequent detection of S. oralis in RAS than in healthy control samples [5]. Cross-reactive recognition of the mycobacterial and human heat shock protein 65-60 antigen by T cells of RAS patients has been reported, suggesting the role of Mycobacterium tuberculosis and autoreactive T cells in RAS [6]. Accumulated evidence supports the association between RAS and Helicobacter pylori infection, but the presence of H. pylori at the RAS lesions is controversial [7, 8]. In an attempt to discover the microorganisms present in RAS lesions, Marchini et al. [9] studied the mucosal microbiota in RAS patients using a culture-independent method. Due to limitations in the methods available at that time, only 57 and 38 phylotypes were defined from 10 RAS and 10 healthy subjects, respectively. Recently, the salivary microbiota in patients with inflammatory bowel disease, where RAS is one of the extraintestinal manifestations of inflammatory bowel disease [10], and the oral mucosal microbiota in RAS patients have been studied by high-throughput sequencing of the 16S rRNA genes [11, 12]. In addition, Terminal-Restriction Fragment Length Polymorphism analysis of bacterial 16S rRNA genes, the human oral microbe identification microarrays, and matrix-assisted laser desorption/ionization time-of-flight analysis have been applied to study the oral microbiota of RAS patients [13, 14]. Although all previous studies suggested that imbalances in the oral microbiota may be involved in the etiopathogenesis of RAS, no specific bacterial species associated with RAS have been identified. We previously characterized the murine oral microbiota to the species level by pyrosequencing [15]. Therefore, this study aimed to characterize the microbiota of the oral mucosa and saliva of RAS patients compared with control subjects at the species level. Pyrosequencing analysis successfully characterized the oral microbiota of RAS patients and identified two species associated with RAS risk.
Results
Subjects
The demographic data of RAS patients and control subjects included in the current study are summarized in Table 1. Nine males and nine females with the active lesions of minor RAS were included and the age distribution of the patients was 19 to 81 years. The mucosal sampling sites of RAS lesions included the tip of the tongue (n = 4), the buccal mucosa (n = 4), and the labial mucosa (n = 10). Twelve patients had a single ulcer and six patients had multiple ulcers on the tip of the tongue, labial mucosa, and soft palate. The control group included eight males and 10 females with an age distribution of 21 to 71 years. The mucosal sampling sites included the buccal mucosa (n = 11) and the labial mucosa (n = 7).
The alpha and beta diversities of the oral microbiota
From the 51 communities, total 484,501 filtered reads (average 9500 reads per sample) with an average length of 479 bp were obtained, which resulted in greater than 99 % Good’s coverage for each sample. We first compared the alpha diversity between the control and RAS group. The species richness of the RAS microbiota estimated by Chao 1 was not significantly different from that of controls either in the mucosa (314 ± 19 vs. 292 ± 22) or in the saliva (377 ± 14 vs. 447 ± 33). The diversities of RAS microbiota determined by the Shannon index were also comparable to those of controls both in the mucosa (3.57 ± 0.11 vs. 3.56 ± 0.06) and in the saliva (4.02 ± 0.09 vs. 4.21 ± 0.1) (Fig. 1a).

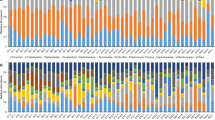
Comparison of mucosal and salivary microbiota between control and RAS. a The species richness estimated by Chao1 and Shannon diversity index are expressed using box and whisker plots. b PCoA plot generated using weighted Unifrac metric. The two components explained 52 % of variance. (unfilled symbols: control samples, filled symbols: RAS samples). c The intra- and intergroup Unifrac distances of mucosal communities were obtained using weighted metric. d Double pie charts present the mean relative abundance of dominant phyla (top 5) and genera (top 15). * denotes significant difference by Mann-Whitney U test (P < 0.01)
UniFrac-based principal coordinate analysis (PCoA) to determine variation among the samples revealed that the microbiota profile was differentiated better by the anatomical sites, i.e., mucosal surfaces vs. saliva, than by disease. The different locations in the mucosa, i.e., tongue tip, labial, or buccal, did not show distinct clustering (Fig. 1b). Although PCoA clustering did not reveal clear separation between the control and RAS communities, the intergroup UniFrac distance (0.061 ± 0.001) was higher than the intragroup distance of controls (0.057 ± 0.001), suggesting a significant difference in the bacterial profile between control and RAS samples. In addition, a higher intragroup UniFrac distance in the RAS (0.067 ± 0.001) compared to the control group suggested the increased inter-subject variability for RAS lesions (Fig. 1c). In the salivary communities that reflect not only the diseased sites but also the healthy sites of patients, no significant differences were observed in intra- or intergroup UniFrac distances.
Differences in oral microbiota composition between the control and RAS group
Next, the relative abundance of each taxon between the control and RAS group was compared. Although a total of 26 different phyla were identified from the mucosa samples, Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteria encompassed the majority of the sequences (>99 % in Controls and >97 % in RAS). The relative abundance of the major phyla observed in the control subjects was not significantly different from that of the RAS patients (Fig. 1d). However, the relative abundance of Streptophyta, a minor phylum, was significantly increased in the RAS group (P = 0.03). At the genus level, Streptococcus constituted almost half of the total mucosal microbiota in the control group and the other major genera included Haemophilus, Rothia, Neisseria, Actinomyces, and Veillonella. Compared to controls, the RAS mucosal communities contained significantly reduced abundance of Veillonella (Fig. 1d). Fourteen other genera also showed differences in the relative abundance (Table 2). At the species level, the abundance of several Streptococcus, including S. salivarius, V. dispar, R. dentocariosa, A. odontolyticus, and Prevotella histicola was decreased in the RAS mucosa. Instead, the abundance of Acinetobacter oryzae, A. johnsonii, Capnocytophaga sputigena, N. oralis, Myxococcus xanthus, Ruminococcus gnavus, and Treponema denticola was increased (Table 2).
The salivary microbiota of RAS tended to contain decreased Firmicutes and increased Proteobacteria among the major phyla, but the differences were not significant. At the phylum level, only SR1 showed a significant difference (0.01 ± 0.004 vs 0.6 ± 0.5, P = 0.02). Among the top 15 genera shown in Fig. 1d, the RAS salivary microbiota was populated by a significantly increased Porphyromonas, and the relative abundance of three other genera was also increased (Table 3). At the species level, the abundance of S. salivarius was decreased in the RAS samples. Instead, the abundance of N. flava, N. sicca, C. gingivalis, C. sputigena, Aggregatibacter segnis, Abiotrophia defectiva, and unclassifieded Porphyromonas species (FM995684_s and Porphyromonas_uc) was increased (Table 3).
Identification of bacterial species associated with RAS
We explored if such changes in the abundance of bacterial species is associated with RAS risk. A logistic regression analysis of the top 100 species in the mucosal microbiota using a forward method revealed that the abundance of S. salivarius was associated with a reduced RAS risk (OR 0.734 per 1 % increase, CI 95 % 0.565-0.954, P = 0.02), and the abundance of A. johnsonii was associated with an increased RAS risk (OR 211 per 1 % increase, CI 95 % 1618-2.7E4, P = 0.03). None of the species in the salivary microbiota showed significant association with RAS. A dysbiosis index was defined as 5.35 × [A. johnsonii] - 0.309 × [S. salivarius] using the relative abundance of A. johnsonii and S. salivarius in the mucosa where 5.35 and -0.309 are the regression coefficients. The dysbiosis index was significantly associated with RAS risk (OR 2.76. CI 95 % 1.26-6.05, P = 0.01) and correctly predicted 83 % of the total cases for the absence or presence of RAS (94 % of control and 72 % of RAS, Fig. 2).

Dysbiosis index of RAS. A dysbiosis index was defined as 5.35 × [A. johnsonii] - 0.309 × [S. salivarius] using the relative abundance of A. johnsonii and S. salivarius in the mucosa where 5.35 and -0.309 are the regression coefficients. The dysbiosis index of 18 control samples and 18 RAS samples are shown. The dotted line indicates cutoff for RAS
To understand the potential role of bacteria in the etiopathogenesis of RAS, the effects of two RAS-associated species on the viability and proliferation of oral epithelial cells were examined. P. gingivalis, a periodontal pathogen that has been reported to inhibit wound healing in an in vitro scratch assay [16], was used as a control. A. johnsonii showed greater cytotoxicity against HOK-16B cells than S. salivarius that showed low levels of cytotoxicity only at MOI 1000 (Fig. 3a). Interestingly, A. johnsonii substantially inhibited the proliferation of HOK-16B cells in a dose dependant manner (Fig. 3b).

The effect of RAS-associated bacterial species on the viability and proliferation of human oral epithelial cells. HOK-16B cells were infected with A. johnsonii, S. salivarius, and P. gingivalis at MOIs of 100, 500, and 1000 for 24. The viability (a) and the number of live HOK-16B cells (b) in six wells from two independent experiments were determined by trypan blue exclusion and compared with control cells without bacterial infection. *: P < 0.05, **: P < 0.01, ***: P < 0.001
Discussion
In this study, we showed that imbalances in the mucosal and salivary microbiota are associated with RAS. Among the top 15 genera observed in the mucosa of control subjects, 13 except for Escherichia and Lautropia were common to the major genera characterized in normal adults enrolled in the HMP. Similarly, 14 genera except for Lautropia out of the top 15 genera observed in the saliva were common to the HMP [17], defining them as the healthy ‘core microbiota’ of oral microbial communities. Although most of the genus members of the normal flora were shared between Koreans and the HMP subjects, the order of the genus composition and the relative abundance of the major phyla were different (Additional file 1: Figure S1).
Among the 15 species/phylotypes significantly decreased in the RAS mucosa compared to the controls, nine species including S. salivarius, S. parasanguinis, S. peroris, S. vestibularis, S. lactarius, V. dispar, Rothia dentocariosa, Campylobacter concisus, Actinomyces odontolyticus, and P. histicola belonged to those defined as the normal flora of the oral mucosa [1, 17], and the six unclassified phylotypes also belonged to Streptococcus and Prevotella. In contrast, the seven species that were significantly increased in the RAS mucosa did not belong to the normal oral mucosal flora. A. oryzae and N. oralis have recently been isolated from wild rice and healthy gingival plaque, respectively [18, 19]. Myxococcus xanthus is a ubiquitous soil bacterium [20]. A. johnsonii is known to be a member of the skin flora [21]. A. johnsonii, the species associated with an increased RAS risk, has been isolated from clinical samples in association with bacteremia [22]. R. gnavus is a member of the gut flora [23]. Interestingly, an increase in R. gnavus in the gut flora is associated with Crohn’s disease [24]. In the salivary microbiota of RAS patients, the decrease in S. salivarius and the increase in Capnocytophaga sputigena were common to the changes observed in the mucosal microbiota. Capnocytophaga is normally found in the oral cavity but considered as an opportunistic pathogen involved in various infections, including endodontic infections, emphysema, and bacteremia [25–27]. Collectively, these results indicate decreases in the members of healthy core microbiota but increases in rare species in the mucosal and salivary microbiota of RAS patients. Particularly, a decrease in S. salivarius and an increase in A. johnsonii in the mucosa were associated with increased RAS risk.
Hijazi et al. reported changes in the relative abundance of only five unclassified genera in ulcerated sites of RAS patients compared with control subjects [12], probably due to the smaller size of the subjects and the smaller number of reads per sample compared to the current study. Because all of the reported genera are unclassified, it is difficult to appreciate the biological relevance of the changes in those. However, the increase of Porphyromonadaceae and decrease of Streptococcaceae in RAS reported by Hijazi et al. may agree with the increase of Porphyromonas in the saliva and decrease of many streptococcal species in the mucosa observed in the current study. The decrease in Streptococcus was also common in two other studies [5, 10]. The decrease of Veillonella in the RAS oral mucosa coincides with the study by Seoudi et al. [14]. The abundance of S. oralis was not different between the control and RAS groups, confirming the lack of association of S. oralis with RAS [5]. Either H. pylori or M. tuberculosis, the species proposed as bacterial etiology of RAS [6, 7], was not detected in any subjects of the current study.
Some diseases are associated with changes in microbial diversity. For example, periodontitis is associated with the increased diversity of plaque bacteria, while Crohn’s disease is associated with the reduced diversity of colonic microbiota [28, 29]. RAS was not associated with changes in the alpha-diversity of the mucosal or salivary microbiota, which agrees with previous studies [11, 13]. However, increased inter-subject variability of the mucosal microbiota was observed in the RAS patients. The increased inter-subject variability was not attributed to the different sampling sites, because the intragroup distance of RAS was still significantly higher than that of control group after removing the tongue tip samples (0.066 ± 0.002 vs. 0.057 ± 0.001, P = 0.0002). A similar high inter-sample variability has been reported in chemotherapy-related oral mucositis lesions [30]. The increased inter-subject variability may underlie the increased number of phylotypes characterized in the RAS mucosa in a study by Marchini et al. in which the samples from 10 RAS patients were pooled [9].
The dysbiosis index developed using the relative abundance of A. johnsonii and S. salivarius correctly predicted 83 % of the total cases for the absence or presence of RAS. This is a cross-sectional study, and there is no evidence that the bacterial species increased at the RAS lesions have a role in the initiation or progression of the disease. However, the lessons from the study of dental caries and periodontitis suggest that the bacterial species increased at the lesions may include the major pathogenic bacteria of the disease. Interestingly, A. johnsonii substantially inhibited the proliferation of gingival epithelial cells and showed increased cytotoxicity against epithelial cells. It has been reported that the relative abundance of Streptococcus was negatively associated with the concentrations of IL-1β and IL-8 in saliva [11]. Therefore, the imbalance of A. johnsonii and S. salivarius could contribute to ulceration, delayed healing, and severe pain caused by inflammatory cytokines, all of which are associated with RAS. Further host cell-microbe interactions are currently being studied.
When the imbalance between the healthy species such as S. salivarius and the potentially harmful species such as A. johnsonii is confirmed as the etiology of RAS, either probiotic application of S. salivarius or antibiotics that selectively kill harmful species but not healthy species may provide a cure for RAS by restoring the balance.
The current study has several limitations. First, the mucosal specimens of the control subjects were sampled from the labial and buccal mucosa, while the sampling sites from the RAS patients also included the tip of the tongue. Second, the limited number of total cases requires further study in larger cohorts and also in diverse populations, considering the differences in the relative abundance of major phyla comprising healthy microbiota between Koreans and the HMP subjects. Third, the non-ulcer sites of RAS patients were not studied. According to the study by Bankvall et al., the mucosal microbiota at the non-ulcer sites of RAS patients was different from that of controls, and the differences were most profound in patients who had lesions during sampling [13]. We originally designed the study to compare the mucosal microbiota in the lesions of RAS patients also with that in the post-healing sites. However, only four patients re-visited the clinic for additional sampling after healing, and those samples were not included in the current study. Longitudinal studies in the future will provide valuable evidence for the role of bacteria in the ediopathogenesis of RAS.
Conclusion
Pyrosequencing analysis successfully characterized the oral microbiota of RAS patients compared with healthy controls at the species level. The mucosal microbiota of RAS lesions are characterized as decreases in the members of healthy core microbiota but increases of rare species, and a decrease in S. salivarius and an increase in A. johnsonii are associated with RAS risk. These findings may provide a diagnostic tool and new targets for the therapeutic management of RAS.
Methods
Ethics, consent and permissions
This study was performed according to the Declaration of Helsinki and conformed to the STROBE guidelines. The protocol was approved by the institutional review board at the Seoul National University Dental Hospital (CRI 12018). Written informed consent was obtained from all subjects.
Study population and sample collection
Twenty patients with minor RAS active lesions who visited the Oral Medicine Clinic at the Seoul National University Dental Hospital from February 2013 to January 2014 and 20 control subjects without oral mucosal disorders were enrolled. Subjects who had received antibiotics or steroid within the last month, and patients with xerostomia (unstimulated whole salivary flow rate <0.1 ml/min) were excluded. Subjects with other oral mucosal diseases (Candida count > 1000 colony forming unit/ml, hematologic deficiency related diseases) or systemic diseases that involve oral ulcers were also excluded. We also compared the microbiota in the saliva of RAS, which reflects the microbiota in both the healthy and diseased sites of patients, with that of control subjects. All subjects were asked to avoid eating and antiseptic mouthwashes for two hours before sampling. For the mucosa sampling, a sterilized 20 mm × 20 mm polyvinylidene difluorid membrane (Korea, Seoul, Korea) was placed on the largest ulcerated area of patients for 30 s. Samples were taken from the labial or buccal mucosa of healthy subjects. A minimum of 2 ml unstimulated whole saliva samples were collected by a spit method.
DNA extraction, 16S rRNA gene amplification, and pyrosequencing
Genomic DNA was isolated from the membranes and the pellets of centrifugated saliva using the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA). Forty mucosal (n = 20 and n = 20 for Controls and RAS, respectively) and 20 salivary (n = 10 and n = 10 for Controls and RAS, respectively) samples were subjected to pyrosequencing analysis. The gDNA was amplified using primers targeting the V1 to V3 hypervariable regions of bacterial 16S rRNA gene, and the PCR products were sequenced according to the previously described method [15] using a 454 GS FLX Titanum Sequencing System (Roche, Branford, CT, USA). Both the 16S rRNA gene amplification and sequencing were performed at ChunLab Inc. (Seoul, Korea). Out of the 60 samples analyzed, we successfully obtained data sets for 39 mucosal (n = 19 and n = 20 for Controls and RAS, respectively) and 17 salivary (n = 8 and n = 9 for Controls and RAS, respectively) microbiota communities. Four samples failed in pyrosequencing due to insufficient amplification of the 16S rRNA genes, although all DNA samples passed the quality control. Because communities from smokers are excluded from the final data set, the current study includes only 36 mucosal (n = 18 and n = 18 for Controls and RAS, respectively) and 15 salivary (n = 7 and n = 8 for Controls and RAS, respectively) microbiota communities. The pyrosequencing data, including those from smokers, are available in the SRP database under the accession number SRP049562. The results of data analysis including smokers are also provided as Additional file 1 (Figs. 2 and 3 and Tables 1, 2 and 3). The entire process from the enrollment of subjects to the acquisition of final data sets is illustrated as a flow chart (Fig. 4).

A flow chart from the enrollment of subjects to the acquisition of final data sets
Pyrosequencing data analysis
The basic analysis was conducted according to previously published descriptions [15]. After removing PCR primer sequences, any reads containing two or more ambiguous nucleotides or reads shorter than 300 bp were discarded. Chimera sequences detected by the Bellerophone method [31] were also removed. The taxonomic classification of each read was assigned against the EzTaxon database-e (http://www.ezbiocloud.net/eztaxon) [32]. The species richness and diversity index were calculated using the Ribosomal RNA database project’s pyrosequencing pipeline (http://pyro.cme.msu.edu). The cutoff value for assigning a sequence to the same phylotype was ≥ 97 % similarity. Random subsampling was conducted to equalize variation in the read counts among the samples. The overall phylogenetic distance between communities was estimated using the weighted Fast UniFrac [33] and was visualized using PCoA. In addition, the pyrosequencing data of buccal mucosa and saliva published by The Human Microbiome Project (HMP) Consortium [34] were also analyzed to compare with the control subjects of the current study.
Bacterial and epithelial cell culture
Because isolation of bacteria from the patients was not included in the original protocol approved by the institutional review board, type strain was used for additional functional study. A. johnsonii KCTC 12405 (Korean Collection for Type Culture, Daejeon, Korea) was cultured in BHI medium at 30 °C and aerobic atmosphere. S. salivarius KCTC 5512 (KCTC) and P. gingivalis ATCC 33277 (American Type Culture Collection, Manassas, VA, USA) were cultured in ATCC medium 188 and BHI medium supplemented with 5 μg/ml hemin and 10 μg/ml vitamin K, respectively, at 37 °C under an anaerobic atmosphere. Bacteria in log phase growth were harvested and bacterial concentrations were determined by flow cytometry [35]. Immortalized human oral keratinocyte HOK-16B cells originated from the retromolar gingival tissue [36] were maintained in keratinocyte growth-culture medium (Clonetics, San Diego, CA, USA) containing supplementary growth factors. HOK-16B cells plated into 48-well plates at 4x104 cells/well in triplicate were cultured for 24 h and then infected with bacteria at the multiplicity of infection (MOI) 0, 100, 500, and 1000 as previously described [37]. After 24 h of co-culture at 37 °C in a water-saturated atmosphere of 95 % air and 5 % CO2, cells were harvested, including the dead cells in the supernatant. The viability and total number of live cells in each well were determined by trypan blue exclusion under a microscope.
Statistics
The data are presented as the mean ± the standard errors of means, unless described otherwise. The differences in relative abundance and in UniFrac distances between the two groups were determined with the Mann-Whitney U-test and t-test, respectively. The association of bacterial species with RAS risk was determined with a logistic regression analysis. Differences in the viability and proliferation between control and infected cells were analyzed by t-test. All statistics were performed using the SPSS Statistics19 software (SPSS Inc., Chicago, IL, USA). Significance was set at P < 0.05.
Abbreviations
- HMP:
-
Human Microbiome Project
- PCoA:
-
principal coordinate analysis
- RAS:
-
recurrent aphthous stomatitis
References
Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–32.
Scully C, Felix DH. Oral medicine--update for the dental practitioner. Aphthous and other common ulcers. Br Dent J. 2005;199:259–64.
Slebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz). 2014;62:205–15.
Hoover CI, Olson JA, Greenspan JS. Humoral responses and cross-reactivity to viridans streptococci in recurrent aphthous ulceration. J Dent Res. 1986;65:1101-4.
Riggio MP, Lennon A, Ghodratnama F, Wray D. Lack of association between Streptococcus oralis and recurrent aphthous stomatitis. J Oral Pathol Med. 2000;29:26–32.
Hasan A, Childerstone A, Pervin K, Shinnick T, Mizushima Y, Van der Zee R, et al. Recognition of a unique peptide epitope of the mycobacterial and human heat shock protein 65-60 antigen by T cells of patients with recurrent oral ulcers. Clin Exp Immunol. 1995;99:392–7.
Adler I, Muiño A, Aguas S, Harada L, Diaz M, Lence A, et al. Helicobacter pylori and oral pathology: relationship with the gastric infection. World J Gastroenterol. 2014;20:9922–35.
Mansour-Ghanaei F, Asmar M, Bagherzadeh AH, Ekbataninezhad S. Helicobacter pylori infection in oral lesions of patients with recurrent aphthous stomatitis. Med Sci Monit. 2005;11:CR576–9.
Marchini L, Campos MS, Silva AM, Paulino LC, Nobrega FG. Bacterial diversity in aphthous ulcers. Oral Microbiol Immunol. 2007;22:225–31.
Vavricka SR, Schoepfer A, Scharl M, Lakatos PL, Navarini A, Rogler G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm Bowel Dis. 2015;21:1982–92.
Said HS, Suda W, Nakagome S, Chinen H, Oshima K, Kim S, et al. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res. 2014;21:15–25.
Hijazi K, Lowe T, Meharg C, Berry SH, Foley J, Hold GL. Mucosal microbiome in patients with recurrent aphthous stomatitis. J Dent Res. 2015;94:87S–94S.
Bankvall M, Sjöberg F, Gale G, Wold A, Jontell M, Östman S. The oral microbiota of patients with recurrent aphthous stomatitis. J Oral Microbiol. 2014;6:25739.
Seoudi N, Bergmeier LA, Drobniewski F, Paster B, Fortune F. The oral mucosal and salivary microbial community of Behçet’s syndrome and recurrent aphthous stomatitis. J Oral Microbiol. 2015;7:27150.
Chun J, Kim KY, Lee JH, Choi Y. The analysis of oral microbial communities of wild-type and toll-like receptor 2-deficient mice using a 454 GS FLX Titanium pyrosequencer. BMC Microbiol. 2010;10:101.
Laheij AM, de Soet JJ, Veerman EC, Bolscher JG, van Loveren C. The influence of oral bacteria on epithelial cell migration in vitro. Mediators Inflamm. 2013;2013:154532.
Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012;13:R42.
Wolfgang WJ, Passaretti TV, Jose R, Cole J, Coorevits A, Carpenter AN, et al. Neisseria oralis sp. nov., isolated from healthy gingival plaque and clinical samples. Int J Syst Evol Microbiol. 2013;63:1323–8.
Chaudhary HJ, Peng G, Hu M, He Y, Yang L, Luo Y, et al. Genetic diversity of endophytic diazotrophs of the wild rice, Oryza alta and identification of the new diazotroph, Acinetobacter oryzae sp. nov. Microb Ecol. 2012;63:813–21.
Vos M, Velicer GJ. Isolation by distance in the spore-forming soil bacterium Myxococcus xanthus. Curr Biol. 2008;18:386–91.
Seifert H, Dijkshoorn L, Gerner-Smidt P, Pelzer N, Tjernberg I, Vaneechoutte M. Distribution of Acinetobacter species on human skin comparison of phenotypic and genotypic identification methods. J Clin Microbiol. 1997;35:2819–25.
Turton JF, Shah J, Ozongwu C, Pike R. Incidence of Acinetobacter species other than A. baumannii among clinical isolates of Acinetobacter: evidence for emerging species. J Clin Microbiol. 2010;48:1445–9.
Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–7.
Willing BPDJ, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–54.
Murad CF, Sassone LM, Faveri M, Hirata Jr R, Figueiredo L, Feres M. Microbial diversity in persistent root canal infections investigated by checkerboard DNA-DNA hybridization. J Endod. 2014;40:899–906.
Li A, Tambyah P, Chan D, Leong KK. Capnocytophaga sputigena empyema. J Clin Microbiol. 2013;51:2772–4.
Gomez-Garces JL, Alos JI, Sanchez J, Cogollos R. Bacteremia by multidrug-resistant Capnocytophaga sputigena. J Clin Microbiol. 1994;32:1067–9.
Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–85.
Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, et al. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn’s but not in ulcerative colitis. Am J Gastroenterol. 2012;107:1913–22.
Ye Y, Carlsson G, Agholme MB, Wilson JA, Roos A, Henriques-Normark B, et al. Oral bacterial community dynamics in paediatric patients with malignancies in relation to chemotherapy-related oral mucositis a prospective study. Clin Microbiol Infect. 2013;19:E559–67.
Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–9.
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK, et al. EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol. 2007;57:2259–61.
Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Shin J, Ji S, Choi Y. Ability of oral bacteria to induce tissue-destructive molecules from human neutrophils. Oral Dis. 2008;14:327–34.
Kim Y, Jo AR, da Jang H, Cho YJ, Chun J, Min BM, et al. Toll-like receptor 9 mediates oral bacteria-induced IL-8 expression in gingival epithelial cells. Immunol Cell Biol. 2012;90:655–63.
Park NH, Min BM, Li SL, Huang MZ, Cherick HM, Doniger J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis. 1991;12:1627–31.
Acknowledgements
This research was supported by a grant from the Seoul National University Dental Hospital Research Fund (No. 01-2013-0003) and a Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A3005669 and 2013070465).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YK analyzed microbiota, performed cellular work, and drafted manuscript. YSC analyzed microbiota and performed the statistical analysis. KJB processed the clinical samples. SY analyzed the data of HMP. HKP participated in the design of study, collected clinical samples and data, and drafted manuscript. YC conceived of the study, participated in its design, analyzed data, and revised manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1:
The comparison of the oral mucosa and saliva microbiota between the control subjects of current study and HMP subjects at the phylum level. (PDF 428 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kim, Yj., Choi, Y.S., Baek, K.J. et al. Mucosal and salivary microbiota associated with recurrent aphthous stomatitis. BMC Microbiol 16, 57 (2016). https://doi.org/10.1186/s12866-016-0673-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-016-0673-z