
Abstract
Background
Hylurgus ligniperda (Coleoptera: Curculionidae) is a worldwide forest quarantine pest. It is widely distributed, has many host tree species, and possesses strong adaptability. To explore its environmental adaptability and the related molecular mechanisms, we conducted chromosome-level genome sequencing and analyzed the transcriptome under different environmental factors, identifying key expressed genes.
Results
We employed PacBio, Illumina, and Hi-C sequencing techniques to assemble a 520 Mb chromosomal-level genome of H. ligniperda, obtaining an N50 of 39.97 Mb across 138 scaffolds. A total of 10,765 protein-coding genes were annotated after repeat masking. Fourteen chromosomes were identified, among which Hyli14 was determined to be the sex chromosome. Survival statistics were tested over various growth periods under high temperature and low humidity conditions. The maximum survival period of adults reached 292 days at 25 °C, 65% relative humidity. In comparison, the maximum survival period was 14 days under 35 °C, 65% relative humidity, and 106 days under 25°C, 40% relative humidity. This indicated that environmental stress conditions significantly reduced adults' survival period. We further conducted transcriptome analysis to screen for potentially influential differentially expressed genes, such as CYP450 and Histone. Subsequently, we performed gene family analysis to gain insights into their functions and interactions, such as CYP450 and Histone. CYP450 genes affected the detoxification metabolism of enzymes in the Cytochrome P450 pathway to adapt to different environments. Histone genes are involved in insect hormone biosynthesis and longevity-regulating pathways in H. ligniperda to adapt to environmental stress.
Conclusions
The genome at the chromosome level of H. ligniperda was assembled for the first time. The mortality of H. ligniperda increased significantly at 35 ℃, 65% RH, and 25 ℃, 40% RH. CYP450 and Histone genes played an important role in response to environmental stress. This genome offers a substantial genetic resource for investigating the molecular mechanisms behind beetle invasion and spread.
Similar content being viewed by others


Background
Hylurgus ligniperda (Coleoptera: Curculionidae) infects newly cut logs and sometimes attacks healthy trees [1] [2]. It is one of the fastest spreading forest pests [3]. In China, H. ligniperda was first reported to infect Pinus thunbergii in Yantai of Shandong province in 2021. The local P. thunbergia forest was also infested by pine wood nematode (PWN) disease. The cutting piles and roots left by the control of pine wood nematode (Bursaphelenchus xylophilus) have intensified the invasion and colonization of H. ligniperda. This complicates the prevention and control efforts in the local forest [4]. H. ligniperda exhibits a complex life history, with different generations in different regions, and the maximum population varies with seasonal weather [5,6,7]. No reports exist on the environmental stress affecting the growth and development of adults or the internal molecular regulatory mechanisms.
Environmental factors have been shown to directly affect the growth and development of insects [8], and environmental stress particularly impacts insect lifespan and development. Enhanced stress resistance and environmental tolerance promote insect growth and invasion [9]. Prior research has indicated that H. ligniperda exhibits a preference for low temperatures and high humidity conditions [2]. There are no reports on how environmental stress impacts H. ligniperda adults and their internal molecular regulatory mechanisms, which may be the key factors influencing their invasive capacity.
Hylurgus ligniperda originated from the Mediterranean coast of southern Europe and northern Africa [10,11,12]. It has effectively broadened its host range to encompass more than 20 tree species across diverse regions, including Japan, Korea, Turkey, Sri Lanka, Australia, New Zealand, Brazil, Chile, Uruguay, and the United States [13,14,15]. As a pioneer species of secondary pests, H. ligniperda has a broad range of host trees [16]. At present, in China, H. ligniperda is primarily colonized in the coastal areas of Shandong Province, including Weihai, Yantai, and Tai'an. [17]. Additionally, it can carry fungi [18] that are lethal to its hosts. It thrives in cool, humid conditions [19], and adults preferentially feed on the rhizomes of 1-2-year-old seedlings [20] [21]. At 25°C, development from egg to adult typically takes about 45 days [22]. In Southeastern France, the insects’ life history exhibits significant variability across environments and years, suggesting a high level of adaptability possibly influenced by diverse regulatory genes [23]. Our research group has discovered that some post-emergence adults can survive for over 292 days at 25°C and 65% relative humidity (RH) (unpublished data), a significantly longer duration than other Curculionidae beetles. Adults inflict prolonged damage on the host, highlighting the need to investigate their adaptation to hosts and environmental tolerance.
Genome sequencing offers extensive data on particular species and is essential for addressing major scientific obstacles in biological study [24]. Insect genome analysis is essential for pest control and investigating internal molecular mechanisms [25]. In the past, sequencing has faced challenges due to technical limitations and high prices. Technological advancements and cost reductions have increased the accessibility of insect genome analysis. Additionally, InsectBase 2.0 (http://v2.insect-genome.com/) serves as a platform that comprehensively covers insect genome information, facilitating the acquisition of genome data. Up until June 2024, more than 503 Coleoptera genomes have been published, of which 46 species belong to Curculionidae, such as Ips typographus [26], Dendroctonus ponderosae [27], Dendroctonus valens [28], Hypothenemus hampei [29], Ips nitidus [30]. H. ligniperda, a major invasive pest, exhibits a wide host range, prolonged periods of damage, and strong environmental adaptability. This pest poses a continuous threat to its host species. Understanding the mechanisms of invasion and environmental adaptability is crucial for mitigating the further colonization of this beetle. Publishing its genome data is crucial for its stress adaptability and molecular mechanism of spreading.
Histone modifications play a role in regulating insect growth. In Drosophila melanogaster, Histones H1, H2A, H2B, H3, and H4 exist in tandem [31]. Histone H4 regulates the growth and development [32]. In insects, the primary function of cytochrome P450 (CYP450) is to participate in the metabolism of internal and external substances and play a role in detoxification [33]. Insect CYP450s have been classified into six CYP families: CYP6, CYP9, CYP12, CYP18, and CYP28 are unique to insects, while the CYP4 sequence is shared between insects and other species [34]. Cyclin-dependent kinases (CDKs) [35] significantly regulate cell apoptosis and differentiation, affecting insect development. In Caenorhabditis elegans and D. melanogaster mitochondria participate in the inhibition of gene synthesis, which influences their growth [36].
This study primarily investigates the tolerance and response of H. ligniperda to environmental stress, which aims to elucidate the molecular mechanisms underlying its adaptation to both host and environmental conditions during invasion and colonization. We utilized PacBio, Illumina, and Hi-C sequencing technologies to obtain a reference genome for H. ligniperda at the chromosome level, with a size of 520 Mb. This genome was successfully assembled into 14 chromosomes, including the identification of the sex chromosome (Hlig14). We constructed the phylogenetic trees for the Histone, P450, and CDK gene families with other insects. To investigate the impact of environmental stress on H. ligniperda adults, we specifically examined the high temperature (35°C, 65%RH) and low humidity (25°C, 40%RH), comparing the survival period and differential gene expression with those at 25°C and 65%RH. This study may have significant implications for understanding the host adaptability of H. ligniperda under environmental stress. Additionally, the chromosome-level genome assembly offers valuable genomic resources to enhance our understanding of the longevity mechanisms in Coleoptera.
Results
Chromosome level genome assembly of H. ligniperda
We employed a hybrid sequencing approach, combining Sequel II (PacBio, USA) and Novaseq 6000 sequencing, which yielded 38 Gb of HiFi reads and 96 Gb of Illumina sequencing data (Table S1). Based on Kmer (K=17) estimation, we estimated the genome size to be approximately 620 Mb, slightly exceeding the size of our assembled genome due to repeat sequence collapse (Figure S1).
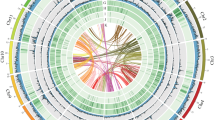
Employing this hybrid sequencing strategy, we assembled the H. ligniperda genome, which exhibited a size of 520 Mb, a GC content of 36%, 145 contigs with an N50 length of 37 Mb, and 138 scaffolds with an N50 length of 39 Mb (Table S2). Following this, 145 contigs were effectively mapped to 14 chromosomes, resulting in a data mapping rate of 96.43% (Figure S2), with only 3.56% remaining as scattered sequences (Figure 1). An assessment of genome completeness and accuracy revealed that reads from HiFi, RNA-seq, and Illumina sequencing all mapped to the assembled genome, with over 95% mapping efficiency (Table S3). Genome completeness was assessed utilizing BUSCO, revealing that complete core genes, including both single-copy and multi-copy genes, represented 98.4%, while the absence of genes comprised merely 0.9%, signifying a notable level of gene integrity (Table S2). In summary, our evaluation results confirm the completeness and suitability of the assembled genome for subsequent analyses.

Genome landscape of Hylurgus ligniperda. From outer to inner circles: (a) Class I TE density; (b) Class II TE density; (c) Density of genes encoding; (d) The proportion of tandem repeat; (e) GC content; (f) Collinear block (Minimum 100 kb). All nonoverlapping Windows are 500 kb in size
Sex chromosome identification
To identify the sex chromosome of H. ligniperda, we performed genome resequencing of sequenced female and male adults, obtaining of 200.2 M and 199.5 M clean reads, respectively. Among the 14 assembled chromosomes using Hi-C (Figure 1), Hlig14 (Chr14) exhibited characteristics consistent with the X chromosome, while the depth ratios of the remaining chromosomes matched those of autosomes (Figure S3). Due to the inherent characteristics of Coleoptera insects, where the Y chromosome is shorter and contains a high degree of repetitive content, we did not assess the presence of a Y chromosome in H. ligniperda, aligning with previous research findings [37].
Genome synteny
We conducted a comparison of chromosome-level genomic synteny involving H. ligniperda, Monochamus saltuarius (Msal10), Tribolium castaneum (Tcas01), Coccinella septempunctata (CSEP10), D. ponderosae (Dpon01) and I. nitidus (Init06). The analysis demonstrated a noteworthy syntenic relationship between chromosome 14 (Hlig14) in H. ligniperda and the X sex chromosomes (Msal10, Tcas01, CSEP10) present in the three other species (Fig. 2). This outcome supports the classification of Hilg14 in H. ligniperda as a sex chromosome, thus aligning with our resequencing results.

Chromosome-scale synteny between H. ligniperda, M. saltuarius, T. castaneum, C. septempunctata, D. ponderosae, and I. nitidus. (a) Whole-genome synteny between H. ligniperda (in color) and M.saltuarius (in black); (b) Whole-genome synteny between H. ligniperda (in color) and T. castaneum (in black); (c) Whole-genome synteny between H. ligniperda(in color) and C. septempunctata (in black). (d) Whole-genome synteny between H. ligniperda (in color) and D. ponderosae (in black); (e) Whole-genome synteny between H. ligniperda (in color) and I. nitidus (in black)
While analyzing for synteny, we discovered many chromosome rearrangement events, indicating extensive differentiation in H. ligniperda. For instance, compared to M. saltuarius, it became evident that each of H. ligniperdas’ chromosomes resulted from the chromosome rearrangements of multiple chromosomes presenting in M. saltuarius. Hyli13 showed synteny with Msal05, Msal04, Msal02, and Msal01, while Hyli14 showed synteny with Msal09, Msal10, Msal02, and Msal01 (Fig. 2a). Similarly, when compared to T. castaneum, Hyli01 was connected to Tcas04 and Tcas02, Hyli09 was from the fusion of Tcas02 and Tcas09, and Hyli10 was connected to Tcas03 and Tcas06 (Fig. 2b). The obvious chromosome rearrangement event was also found in C. septempunctata by comparison, Hlig14 showed synteny with CSEP01, CSEP02 and CSEP10 (Fig. 2c). When compared to D. ponderosae, we found that Hlig14 originated from the fusion Dpon01, Dpon04, Dpon05 and Dpon06; Hlig12 resulted from Dpon05, Dpon07, Dpon09 and Dpon11. In I. nitidus, we found that Hlig14 was derived from Init01, Init02, Init03, and Init06. Therefore, we proposed that the increased number of autosomes in H. ligniperda, primarily stems from chromosome rearrangement events in contrast to the other species.
We further analyzed the presence of collinear CYP450, Histone, and CDK genes on the chromosomes. The analysis revealed the presence of 17 collinear CYP450 genes, 9 CDK genes, and 1 Histone gene between the two species. In contrast, high collinearity for these genes was not observed in other species, thereby underscoring the close genetic relationship between H. ligniperda and D. ponderosae (Fig. 3).

Syntenic analysis of CYP450, Histone, and CDK genes between H.ligniperda and D. ponderosae. H.ligniperda ( in orange); D. ponderosae ( in blue)
Genome annotation
We have successfully identified 60.4% of the repeat elements within the genome of H. ligniperda. Among these repeats, DNA transposons were the most prevalent, constituting 33.66% of the repeating element superfamily, followed by LTR (9.28%), LINE (2.25%), TIR (1.08%), and MITE (0.72%) categories (Table S4). After eliminating duplicate elements, we were able to annotate 10,765 protein-coding genes, which was fewer than the number of genes annotated in most sequenced Curculionidae species. Our assessment of the integrated annotated proteins using BUSCO revealed that 97.1% of the core genes were intact, comprising 96% single-copy and 1% duplicate genes. Furthermore, 97.03% of the coding genes obtained annotations from various databases, including G0 (56.21%), KEGG (34.73%), NR (93.21%), Swiss_Prot (66.93%), Pfam (81.56%), PRINTS (19.55%), CDD (37.14%), TrEMBL (74.18%), and SMART (39.61%) (Table S5).
Phylogenetic analysis of H. ligniperda and 12 other insect species
We obtained genome data for each species from the NCBI (https://www.ncbi.nlm.nih.gov/). To investigate the phylogenetic relationship between H. ligniperda and 12 other insect species, we employed a set of 1630 homologous genes. H. ligniperda and the other nine Curculionoidea insects clustered together to form sister relationships. Among them, H. ligniperda was highly related to D. ponderosae, and relatively distant from Anoplophora glabripennis, T. castaneum, and D. Melanogaster. Our analysis of gene family divergence unveiled that H. ligniperda and D. ponderosae diverged approximately 33.8 (25.4-43.3) Mya. The separation of Curculionoidea and Cerambycidae dated back to roughly 155.3 Mya (147.6-163.5 Mya), while Tenebrionoides’ differentiation occurred around 184.5 Mya (175.9-189 Mya). The divergence of Coleoptera as a whole was estimated at around 298.1 Mya (277.4-342.4 Mya) (Fig. 4), corroborating previous research findings [38].

Species divergence time and gene family expansion and contraction. Numbers at nodes are estimated divergence times with 95% confidence intervals, and pie charts show the proportion of gene families that expanded (green) and contracted (red)
Gene family expansion and contraction
Based on the ultrametric tree and the 9851 homologous genes in 12 species, we used CAFE (v4.2) to detect expanding and contracting gene families. Compared to the common ancestor of H. ligniperda and D. ponderosae, H. ligniperda has 949 contracted genes and 128 expanded genes (Fig. 4; Figure S4). This result indicated that H. ligniperda predominantly underwent gene family contractions rather than expansions during adaptive evolution. To determine the presence or absence of polyploidy and establish ploidy levels, we examined collinear dot plots alongside Ks values. The results consistently demonstrated a 1:1 dot plot ratio across the entire Coleoptera species, signifying the absence of whole-genome duplication events (Figure S5).
Evolution of Histone, CYP450 and CDK gene families in H. ligniperda
In H. ligniperda, we identified 11 Histone genes (Fig. 5) categorized as H1(2 genes), H2A (5 genes), H2B (1 gene), H3(2 genes) and H4(1 gene). The gene family demonstrated notable conservation among diverse species, maintaining consistent domain features. Our analysis revealed no discernible contractions or expansions within the Histone gene family of H. ligniperda. In addition, we identified a total of 60 CYP450 genes (Fig. 6), distributed among CYP6 (12 genes), CYP9 (8 genes), CYP3 (5 genes), CYP4 (21 genes), CYP2 (7 genes) and mitochondrial CYP450 clade (7 genes). Mitochondrial CYP and CYP2 subfamilies, typically conserved and involved in ecdysone or juvenile hormone synthesis or degradation, did not exhibit significant expansions or contractions in H. ligniperda. Conversely, H. ligniperda demonstrated a substantial expansion in other CYP450 subfamilies compared to M. Saltuarius, which possessed only 33 CYP450 genes. Our study found that the number of CYP450 genes in H. ligniperda is much less than 143 CYP450 genes in T. castaneum. Lastly, we identified 14 CDK genes (Fig. 7) and categorized them into four groups based on sequence similarity, with no significant contractions or expansions observed (Table S6).

Maximum-likelihood (ML) phylogenetic tree of Histone genes in the genome of H. ligniperda (node color in purple), A. glabripennis (node color in red), D. ponderosae (node color in dark blue), H. axyridis (node color in orange), H. hampei (node color in green), I. nitidus (node color in light blue) and T. castaneum (node color in pink)

Maximum-likelihood (ML) phylogenetic tree of P450 genes in the genome of H. ligniperda (node color in blue), M. saltuarius (node color in red), T. castaneum (node color in orange)

Maximum-likelihood (ML) phylogenetic tree of CDK genes in the genome of H. ligniperda (node color in purple), A. glabripennis (node color in red), D. ponderosae (node color in dark blue), H. axyridis (node color in orange), H. hampei (node color in green), I. nitidus (node color in light blue), T. castaneum (node color in pink)
Impacts of environmental stress on the survival periods of adult H. ligniperda
We assessed the survival rate of H. ligniperda adults under various environmental stress conditions. The optimal conditions for adult survival were determined at 25 ℃ and 65% relative humidity (RH), with the maximum observed survival periods reaching 292 days. A significant reduction in adult survival periods was observed in the high-temperature group (35 ℃); the survival rate of the high-temperature stress group declined to 0% within 14 days. In the low-humidity group (40%RH), The survival rate decreased to 0% after 106 days (Fig. 8).

Survival statistics under environmental stress conditions of H. ligniperda
Analysis of differentially expressed genes under different temperature and humidity conditions
512 million Illumina reads were generated in RNA sequencing (RNA-seq) data. The clean reads revealed that all samples had a Q20 score exceeding 96% and a Q30 score exceeding 90%. The alignment rate of the clean reads exceeded 84%. These results confirmed the quality of the transcriptome data, qualifying it for subsequent analysis (Table S7).
A total of 5042 differentially expressed genes (DEGs) were obtained. Under high temperatures (Fig. 9a), we observed 1300 up-regulated DEGs and 2166 down-regulated DEGs. Under low humidity conditions (Fig. 9b), there were 829 up-regulated genes and 747 down-regulated genes.

(a) Number of genes between the high temperature and control groups. (b) The number of different genes between the low humidity group and control group. (c) KEGG enrichment analysis of differential genes under high temperature stress. (d) KEGG enrichment analysis of differential genes under low humidity stress
The KEGG functional enrichment analysis was conducted for the DGEs across high temperatures and low humidity. In the high-temperature group, DEGs were predominantly enriched in the Exosome pathway, Lysosome-related pathway, Insect hormone biosynthesis, Glutamate metabolism, and Cytochrome P450 pathway (Fig. 9c). In low humidity, DEGs are primarily concentrated in pathways related to the Chromosome and associated proteins pathway, Cilium and associated proteins, Longevity regulating pathway and the P53 signaling pathway (Fig. 9d). Cytochrome P450 pathway and Chromosome and associated proteins pathway were associated with adult survival, indicating that high temperature and low humidity environmental stress conditions affected those two pathways of H. ligniperda.
We further analyzed the DEGs in the Cytochrome P450 pathway. In the high-temperature group (Fig. 10a), all 17 P450 genes were down-regulated. In the low humidity group (Fig. 10b), we identified three up-regulated CYP450 genes (CYP49a1, CYP345e2, CYP306a1). The detoxification metabolism is facilitated by the expression of CYP450 genes within the Cytochrome P450 pathway. It is thus suggested that H. ligniperda may up-regulate the Cytochrome P450 pathway in response to unfavorable environmental conditions, potentially bolstering its capacity to mitigate harm.

(a) Expression of P450 and Histone genes in the high-temperature group of the transcriptome. (b) Expression of P450 and Histone genes in the low-humidity group of the transcriptome. (c) RT-qPCR analysis of differentially expressed genes at high temperatures. (d) RT-qPCR analysis of differentially expressed genes in low humidity. In all figures, red indicates upregulation, and blue indicates downregulation
Histone genes are notably abundant within the Chromosome and associated proteins pathway and Longevity regulating pathway. In the high-temperature group (Fig. 10a), two Histones (histone-lysine N-methyltransferase 2C-like isoform X2 and histone-lysine N-methyltransferase NSD2) were up-regulated.
In the low humidity group (Fig. 10b), nine Histone genes were down-regulated. The Longevity regulating pathways’ activity in the low-humidity group led to decreased adult survival durations and increased mortality rates. Meanwhile, their up-regulation under high temperatures supports the involvement of Histone genes in insect hormone biosynthesis, which may accelerate the mortality process.
We speculated that CYP450 genes and Histone genes played an important role in the stress resistance to environments in H. ligniperda, to better adapt to the environment and speed up the invasion and colonization.
RT-qPCR was performed to validate the transcriptome analysis and the expression patterns of those key genes. Thirteen crucial DEGs associated with CYP450 and Histone genes were selected (Table S9-S10; Figure S6). Compared with the control group, CYP450 exhibited significant down-regulation in the high-temperature group, and showed up-regulation in the low-humidity group. Histone showed significant up-regulation in the high-temperature and exhibited a down-regulation in the low-humidity group. The obtained results align closely with our transcriptome data, confirming its reliability. It is indicated that under high-temperature conditions, H. ligniperda will up-regulate Histone genes to resist stressful environments, while under low-humidity conditions, it will up-regulate CYP450 genes to adapt to adverse environments. (Fig. 10c-d).
Discussion
Achieving a high-quality genome assembly at the chromosomal level is fundamental for addressing significant biological questions. It facilitates our comprehension of species-level molecular differences [39]. Comparative genomics is pivotal in enhancing our understanding of the timing and mechanisms underlying species divergence [40]. Currently, numerous chromosomal genomes of Coleoptera have been elucidated in prior studies. This wealth of data holds immense value for elucidating the mechanisms underlying species invasions [41]. Previous studies have evaluated the Curculionidae genome more by measuring its size and chromosome count. In the investigation of Scolytidae, D. valens elucidated the population genetic structure via genomic analysis. D. ponderosae identified sex chromosomes from scaffolds. The publication of the genome of I. typographus unveiled expanded gene families linked to plant cell wall degradation, such as pectinases, aspartyl proteases, and glycosyl hydrolases. The genome analysis of H. hampei unveiled a diminished chemosensory receptor gene repertoire and a genome sequence specific to males. The chromosomal-level genome assembly of I. nitidus offers evidence of the species' potential adaptability to conifer hosts and high-altitude hypoxia, and sheds light on the role of fungal symbionts in facilitating these adaptations. We assembled a 520Mb genome with 14 chromosomes and successfully identified the sex chromosome, offering a foundation for the potential identification of additional sex chromosomes in other Curculionidae species. While we did not identify a Y chromosome, its absence in our study does not conclusively negate its existence. Sex is mostly determined by sex chromosomes. The sex determination mechanism and sexual development of insects can be determined by the hierarchical control of several sex determination genes or by symbiotic microorganisms in the body [42, 43]. The curculionidae include diploid species, haplodiploid species, and PGE species [44]. In D. ponderosae, the karyotype is 11 AA + neo-XY. The largest chromosomes in D. ponderosae are the neoX and neo-Y sex chromosomes[45]. Therefore, additional research on H. ligniperda is necessary in the future.
In insects, factors such as dietary restriction (CR) [46], temperature [47], humidity [48], and light [49] have received extensive attention in studies related to growth and development. The growth of Diaphorina citri [50] was shortened by 74% at 41 ℃. Musca domestica [51] activated the oxidative stress process with increased temperature, leading to a shorter lifespan. Studies on Cotesia flavipes [52] have shown that lower humidity, under constant temperature, resulted in a shorter lifespan. Apolygus lucorum [53] has shown adverse effects of low humidity on both survival and oviposition.
Our observations have shown a significant reduction in survival rates when subjected to environmental stress conditions featuring high temperatures and low relative humidity. Comparing living adults with dying or dead adults and linking the up- or down-regulated genes to host adaptation or stress tolerance may present a problem. To address this limitation, future research could focus on longitudinal studies that monitor gene expression changes in H. ligniperda adults under controlled environmental stress conditions over time, allowing for a more precise correlation between gene regulation, host adaptation, and stress tolerance. Our study aimed to examine the effects of high temperature or low humidity conditions on adult survival and compare differentially expressed genes (DEGs) among those conditions. Consequently, we gathered specimens after 14 days of exposure to elevated temperature stress to generate more prominent results. As a close relative of H. ligniperda, D. ponderosae not only exhibits strong chromosomal collinearity, but also shows a high degree of homologous collinearity in CYP450, Histone, and CDK genes, further substantiating the high degree of affinity between the two species.
CYP450 expression patterns vary among different tissues and under various environmental conditions [54]. Different CYP450 subfamilies have been identified in Ips paraconfusus [55] and D. ponderosae [56]. The expression of these genes differed according to sex, gut, and antenna. Most of them were involved in the detoxification of compounds emitted by the host [57]. The CYP450 gene affects bee detoxification and prolongs infected bees' lifespan [58]. Due to the global distribution and broad host adaptability of H. ligniperda [59], our primary focus was analyzing its ability to withstand environmental stress. Our study relied on genomic analysis to provide a more comprehensive and precise assessment of H. ligniperdas’ invasion and colonization under environmental stress conditions. Our findings revealed down-regulation of CYP450 in the high-temperature group and up-regulation in response to low-humidity stress conditions. We hypothesize that adverse environmental conditions may affect CYP450 enzyme activity in H. ligniperda tissues, thereby influencing their stress response. Histone genes control the cell cycle and significantly influence insect growth and development [31]. The homologous gene CpBD-H4 of Histone H4 can enhance the pathogenicity of bacteria in T. castaneum, thereby accelerating mortality [60]. The loss of D. melanogaster Cyclin-dependent protein kinase complex cyclin D/cyclin-dependent kinase4 (CycD /Cdk4) [61] can also shorten lifespan by affecting oxidative stress. Therefore, identifying these three gene families can contribute to a better understanding of the response mechanism underlying host adaptability and environmental stress in H. ligniperda.
Our observations indicate that Histone expression remains stable in H. ligniperda but is significantly up-regulated in response to high temperatures and down-regulated in low humidity conditions. This suggests a correlation between the activity of these genes and the decreased survival rate under environmental stress.
Conclusion
We utilized PacBio and Illumina sequencing platforms, combined with Hi-C library construction, to assemble the H. ligniperda genome at the chromosome level, achieving an assembly size of approximately 520 MB. Based on the scaffold, we successfully assembled 14 chromosomes and identified the sex chromosome. Genomic synteny analysis revealed numerous chromosome fusion events in H. ligniperda. Its CYP450 genes exhibited significant expansion compared to M. saltuarius, possibly due to its wide range of host tree species and polyphagous characteristics. Through transcriptome analysis under different environmental conditions, we investigated the molecular changes in the response of the H. ligniperds to high temperature and low humidity. CYP450 and Histone genes were associated with temperature and humidity stress response.
This work advances future research on the H. ligniperds’ molecular mechanisms and enriches the Coleoptera genome database. Furthermore, this study is instrumental in enhancing our understanding of H. ligniperdas’ host adaptability and environmental stress tolerance.
Materials and methods
Insects
We collected H. ligniperda samples from the roots of P. thunbergii near the Jiuguan coast protection forests in Yantai, Shandong Province, China (37.457 241°N, 121.851 217°E). We performed genome sequencing and Hi-C sequencing analysis on male adult samples. We conducted a re-sequencing analysis on both male and female adult samples. Furthermore, we collected larvae, pupae, females, and males, and treated them under different conditions for transcriptome sequencing. Following sampling, all specimens were promptly frozen in liquid nitrogen and stored at -80°C.
Survival statistics of H. ligniperda adults under different environments
We collected newly emerged adults (with brown elytra) from the roots of P. thunbergii and fed them with phloem for 1 to 2 days until the elytra became completely black. We put emerging adults in a well-ventilated container (Modele: QHP-70BE) and fed them with fresh bark. The food source was changed every two days. The bark was stored in vacuum-sealed bags in the freezer at -20 ℃ to maintain moisture and thawed before feeding. The control group was kept in the dark at a temperature of 25 ℃, 65% RH. In the environmental stress group, we exposed the specimens to dark conditions with high temperature (35 ℃, 65% RH) and low humidity (25 ℃, 40% RH). We placed 30 adults in each group, with three replicates established for each treatment. We replaced the fresh bark every two days and counted the number of surviving adults. At 14 days in the high-temperature group and 48 days in the low-humidity group, we collected three samples from each treatment group and stored them for subsequent analysis.
Genome sequencing
K-mer assessment of genome size
The specific process for DNA extraction is as follows: 1) Take an adult and grind it into powder with liquid nitrogen; 2) Add 1.2ml TNES Buffer, disperse, and evenly divide into 4 1.5ml tubes, and add 100μl TNES Buffer to each tube; 3) Add 10μl Proteinase K(20mg/ml)/tube, and incubate at 58 °C for 6h (PK added once at 2h); 4) At the end of incubation, 375ul 2M NaCl was added to precipitate the proteins; 5) After centrifugation at 1400 RPM for 5min at room temperature, the supernatant was transferred to a new tube and evenly divided into 8 tubes; 6) Add two volumes (700μl) of pre-cooled anhydrous ethanol into each tube and let it stand at -80 ℃ for 30min; 7) Centrifugation at 4 °C for 30 min at 14000rpm, then remove the supernatant (actual: centrifugation for 6min at -20 °C overnight, then centrifugation for 20min under the same conditions the next day); 8) Pre-cooled 70% ethanol, 1 ml was washed for 2 times, centrifuged at 14,000rpm for 10min at 4 ℃; 9) Add 15μl NF.H2O to each tube, stand at 50 ℃ for 1h, and then dissolve. Finally, agarose gel electrophoresis (Tanon, EPS600) was used to detect the concentration. After obtaining sufficient gDNA, 500-1000 ng was fragmented, followed by two-step magnetic bead fragment screening. The Illumina Hiseq platform was employed for library construction, including end repair, ligation, and circularization of selected 80-200 ng fragments with 400 bp sizes. The Qubit® ssDNA Assay Kit was utilized to quantify the purified product. We employed Jellyfish (v2.2.10) [62] and GenomeScope (v2.0) [63], along with the GCE [64] software, to estimate the genome size, heterozygosity, and duplication. Specifically, the genome size was estimated using findGSE [65].
Library construction and sequencing
We sequenced the complete genome of H. ligniperda using PacBio HiFi and Illumina Novaseq 6000 sequencing platforms. We quantified the extracted nucleic acid concentration using Nanodrop (Thermo Fisher Scientific, Nanodrop 2000) and Qubit (Invitrogen, QubitTM3Flurometer). We assessed the integrity of the extracted nucleic acid through agarose gel electrophoresis. The Hi-C library was prepared as follows: tissues were ground with liquid nitrogen, and cross linked with 40 ml of 2% formaldehyde solution for 15 min at room temperature under vacuum, which was inhibited by adding 4.324 ml of Glycine (2.5 M). The cell nucleus was washed with 0.5mL of restriction enzyme buffer, and chromatin was dissolved with diluted SDS. SDS was removed by desiccation with Triton X-100 overnight, and DNA ends were labeled with biotin-14-dCTP. DNA was purified using phenol-chloroform extraction. The Hi-C library was amplified by 12-14 PCR and sequenced using an Illumina HiSeq instrument with a reading of 2×150 bp.
Genome assembly and assessment
Before assembly, we used NextDenovo (https://github.com/Nextomics/NextDenovo) to assemble PacBio HiFi reads into contigs for further analysis. We employed Juicer [66] to align Hi-C reads with the core genome. 3d-DNA was utilized for initial Hi-C supplementary chromosome assembly [67], followed by manual inspection and adjustment using Juicebox [68]. Finally, 3d-DNA was used for re-scaffolding to generate the final draft chromosome genome, gaps of 100 bp were filled using LR_Gapcloser [69] software, and mitochondrial genomes were assembled using GetOrganelle [70]. Using Illumina short reads, we performed two rounds of polishing with Nextpolish [71]. Redundant [72] was employed to align fragmented contig sequences with the chromosome and organelle genome sequences, identifying fragments with low redundancy and coverage for further assembly optimization.
To assess the integrity and accuracy of the H. ligniperda genome assembly, we utilized Burrow-Wheeler Aligner (BWA) (v0.7.17-r1198-dirty) to align the second-generation sequencing data to the genome [73]. Then, we mapped PacBio HIFI reads to the genome using Minimap2 [74]. We mapped RNA-Seq reads to the genomes using HISAT2 [75] filtering out non-primary alignments, and calculated mapping proportion and coverage percentage. Furthermore, we used BUSCO (v5.1.2) [76] to assess the genome assembly quality.
Sex chromosome identification
We identified the sex chromosomes of H. ligniperda by resequencing male and female adults using the Illumina Novaseq 6000 Platform. We employed the BWA (v0.7.17-r1198-dirty) to map the sequenced clean reads to the genome. We calculated the coverage of reads for each chromosome using windows (Bin=50k bp, step=5k bp) with Samtools (v1.15) [77]. Furthermore, we assessed the sequencing coverage for male and female samples by calculating log2 (M/F reads coverage) to identify sex chromosomes. The sequencing coverage of autosomes is identical in male and female samples. The sequencing depth of the X chromosome in the female sample is twice that in males, the Y chromosome is present at higher levels in males.
Annotation of repetitive sequences and non-coding RNA
The transposable element (TE) library was constructed through de novo identification of TEs employing EDTA [78], Repeat Masker (http://www.repeatmasker.org/RepeatMasker/) was applied for recognizing repetitive regions within the genome, tRNAScan-SE [79] for tRNA annotation, Barrnap (https://github.com/tseemann/barrnap) for rRNA annotation, and RfamScan [80] for comparative annotation of diverse non-coding ncRNAs..
Gene prediction and annotation
We utilized 192618 non-redundant protein sequences from all beetles in NCBI RefSeq (https://www.ncbi.nlm.nih.gov/refseq/) database as homologous proteins for H. ligniperda gene annotation. We used MAKER2 [81], EVidenceModeler (EVM) [82], and PASA to generate consistent gene annotations. TEsorter [83] was utilized to detect TE protein domains in the genome. These domains were subsequently masked within EVM. Redundancies in annotations were consolidated, and coding and non-coding genes were uniformly named.
We accessed the integrated annotated proteins using BUSCO (v5.1.2)and annotated the functions of protein-coding genes using three strategies:1) eggNOG-Mapper annotation [84]. We used GO (https://geneontology.org/) and KEGG (https://www.genome.jp/kegg/) to annotate the functions of protein; 2) Sequence similarity search: we employed diamond [85] to compare protein sequences with protein databases (Swiss_Prot, TrEMBL, NR); 3) Domain similarity search: we utilized InterProScan [86] to compare proteins with PRINTS, Pfam, SMART, PANTHER, CDD and other sub-databases to obtain information on amino acid conserved sequence, motifs and domains.
Gene evolution and phylogenetic tree analysis
The phylogenetic tree was constructed by combining protein-coding genes from H. ligniperda and nine other species belonging to the Curculionidae family: D. ponderosae, H. hampei, I. typographus, Listronotus bonariensis, Pissodes strobi, Rhynchophorus ferrugineus, Sitophilus oryzae, E. kamerunicus, I. nitidus. Genome data for all species were obtained from InsectBase 2.0. We performed alignment using 1630 single-copy orthologs obtained through OrthoFinder 2 (v2.0) [87]. We used ModelFinder [88] in IQ-TREE [89] to determine the best model as JTT+F+R4 with bootstrap 1000 times. Subsequently, IQ-TREE was used to construct the phylogenetic tree using the maximum likelihood method.
To estimate species divergence times, we used the MCMCTree from the PAML (v4.9e) [90]. The molecular clock was set to independent rates (clock=2), and the GTR nucleic acid substitution model was used. The first 500,000 rounds of the MCMC chain were set as burn-in. Samples were then taken every 100 rounds, totaling 100,000 samples. To perform a secondary calibration, we used TimeTree (http://www.timetree.org/) to estimate Million Years Ago (Mya) units. Based on the ultrametric tree and the orthologous gene families, we utilized CAFÉ (v4.2) [91] to detect expansions and contractions in gene families. We obtained collinear orthologous gene pairs using MCScanX_h [92]. MUSCLE [93] was used to align protein sequences, PAL2NAL [94] was used to transform into codon alignment, and KaKs_Caculator [95] was used to calculate Ka, Ks, etc.
Chromosome synteny analysis
Genetic comparisons were conducted between H. ligniperda, M. saltuarius, T. castaneum, C. septempunctata, D. ponderosae and I. nitidus using Lastz (https://github.com/lastz/lastz). Collinear visualization was achieved using Circos [96] software. Chromosome assembly data for the remaining five species were sourced from InsectBase 2.0 (http://v2.insect-genome.com/).
Gene family analysis
We retrieved reference protein sequences for the Histone, CDK and P450 families from the InsectBase 2.0 database and utilized them as query sequences. First, we utilized Blastp (diamond blastp -d DATABASE -q INPUT -o OUTPUT --more-sensitive -p 1 --quiet -e 0.001 --compress 1) to search for candidate genes within the genomes of seven species: A. glabripennis, I. nitidus, T. castaneum, H. hampei, H. ligniperda, Harmonia axyridis, and D. ponderosae. Next, we used Hmmsearch software to identify candidate sequences associated with P450, Histone and CDK. Hidden Markov models (HMM) of P450 (PF00067.25) and Histone (PF00125.27) gene families were obtained from the Pfam database. Based on the existing protein sequences, we constructed the HMM of CDK by Hmmbuild program (http://www.hmmer.org/). The candidate sequences obtained through both methods were combined and compared with the NR database. Sequences annotated as P450, Histone and CDK were retained as reference proteins for homology prediction by Genewise (https://www.ebi.ac.uk/~birney/wise2/) software. The gene model was refined through homologous annotation, and redundancy was eliminated to obtain a comprehensive set of gene family members.
We conducted multiple sequence alignments for each gene family using MUSCLE (v3.8.1551) with default software parameters. Then, we employed IQ-TREE software to construct phylogenetic trees using the maximum likelihood (ML) method.
Transcriptome data analysis
Transcriptome Sequencing and evaluation
We selected three adult samples from each group for RNA extraction. The specimens were immersed in a grinding solution and meticulously dissected with sterilized scissors prior to RNA extraction. RNA extraction from all experimental samples was carried out using the RNAprep Pure Tissue Kit (Tiangen, China). We assessed RNA integrity and contamination levels using agarose gel electrophoresis and examined RNA purity with Nanodrop. Quality control for Raw Data, including sequencing errors and GC content analysis, was performed using Fastp [97] software. Transcriptome data quality was evaluated with RSeQC [98] software. Stringtie [99] software was used for splicing.
Gene expression analysis
Gene expression levels were estimated as transcripts per million reads (TPM) using the RSEM (v 1.3.1) [100]. We identified 60S ribosomal protein L13 as a reference gene. Differentially expressed genes (DEGs) between each two groups were identified using the DESeq2 R package (v 1.24.0) and are presented as log2(Fold Change (FC)) values. The p values were adjusted using the Benjamini-Hochberg method [101]. The genes satisfying |log2(FC) |>1 and P ≤0.05 were considered as DEGs. Functional enrichment of DEGs was performed through GO and KEGG.
Real-Time Quantitative PCR Analysis
Real-time quantitative PCR (RT-qPCR) was used to measure the expression patterns of DEGs related to host adaptation and environmental stress tolerance in H. ligniperda. Specific primers for the DEGs and reference genes were designed using Primer3Plus (https://www.primer3plus.com/). We selected genes with TPM > 20, Foldchange < 1; stable expression changes across treatments, and an SD value < 1 as reference genes in various transcriptome treatment groups [102].
The RT-qPCR was performed with the TB Green® Premix Ex Taq™ II (Takara, Japan) and ran on the Bio-Rad CFX96 PCR System (Bio-Rad, USA). Each 25 μL amplification reaction contained 12.5 μL of TB Green Premix Ex TaqII (2×), 1.0 μL of each positive and negative primer (10 μmol/L), 1 μL of cDNA template, and 9.5 μL of ddH2O. The amplification protocol included pre-denaturation at 95°C for 30 seconds, denaturation at 95°C for 5 seconds, and annealing at 60°C for 30 seconds, repeated for 40 cycles with three technical replicates per sample. Gene relative expression was calculated by 2–ΔΔCt method [103].
Statistical analysis
Data were collected and analyzed using Excel 2018 and SPSS 26.0 software (IBM, USA). The longevity of adult beetles under diverse environmental conditions was assessed using normal distribution analysis, followed by subsequent application of one-way ANOVA after confirming conformity to normal distribution. Differences among multiple datasets were evaluated using the Duncan test. Mortality graphs, bar charts, and heat maps were generated using GraphPad Prism 8 software (GraphPad Software, USA).
Data availability
All data mentioned in this paper are available at the National Center for Biotechnology Information (NCBI) with the BioProject no. PRJNA1065270. Raw sequence data (Illumina, Pacbio, Hi-C, resequencing and RNA-seq) have been deposited in the NCBI Sequence Read Archive (SRA) database at the BioSample no. SAMN39456303-SAMN39456311. RNA-seq sequencing data under different environmental conditions have been transmitted at BioSample no. SAMN39456420-SAMN39456431. The annotated GFF file of the genome has been deposited in the Open Science Framework (https://osf.io/7vwnj/?view_only = e350ce100de744ebbab8c6ba4c02c76a).
References
Reay SD, Walsh PJ. Observations of the Flight Activity of Hylastes Ater and Hylurgus ligniperda (Curculionidae: Scolytinae) in Pinus Radiata Forests in the Central North Island. New Zealand New Zealand Entomologist. 2001;24:79–85.
Reay SD, Walsh PJ. Relative Attractiveness of Some Volatiles to the Introduced Pine Bark Beetles, Hylastes Ater and Hylurgus ligniperda (Curculionidae: Scolytinae). New Zealand Entomologist. 2002;25:51–6.
Lin W, Park S, Jiang ZR, Ji YC, Ernstsons AS, Li JJ, Li Y, Hulcr J. Native or Invasive? The Red-Haired Pine Bark Beetle Hylurgus ligniperda (Fabricius) (Curculionidae: Scolytinae) in East Asia. Forests. 2021;12(7):950.
Ren LL, Tao J, Wu HW, Zong SX, Wang CZ, Hua DE, et al. The first discovery and infection characteristics of the major pest Hylurgus ligniperda in China. Forestry science. 2021;57:140–50.
Clare GK, George EM. Life Cycle and Massrearing of Hylurgus Ligniperda Using a Novel Eggcollection Method. New Zealand Plant Protection. 2016;69:143–52.
Ciesla WM. Pine Bark Beetles: A New Pest Management Challenge for Chilean Foresters. J Forest. 1988;86:27–31.
Fabre JP, Carle P. Contribution à l’étude biologique d’Hylurgus Ligniperda (Coleoptera: Scolytidae ) dans le Sud-est de la France. Ann Sci For. 1975;32:55–71.
Sarup P, Sørensen P, Loeschcke V. The Long-Term Effects of a Life-Prolonging Heat Treatment on the Drosophila Melanogaster Transcriptome Suggest That Heat Shock Proteins Extend Lifespan. Exp Gerontol. 2014;50:34–9.
Carey JR. Demographic Mechanisms for the Evolution of Long Life in Social Insects. Exp Gerontol. 2001;36:713–22.
Serez M. Verwendung des aggregationspheromon-Präparats “Ipslure”gegen den mediterranen Kiefernborkenkäfer, Ips (Orthotomicus) erosus (Woll) (Col., Scolytidae). Anz. Schadlingskde, Pflanzenschutz. 1987; 60: 94–95.
Sangwook P, Jong J, Taeman H. A New Species and Five Newly Recorded Species of Scolytinae (Coloptera: Curculionidae) from Korea. Taiwania. 2021;66(1):1–10.
Browne FG. Pests and Diseases of Forest Plantation Trees: An Annotated List of the Principal Species Occurring in the British Commonwealth. Forest & Conservation History. 1969;13:64.
Kateryna D, Rimvydas V, Valentyna M, Audrius M. Fungi Associated with the Red-Haired Bark Beetle, Hylurgus ligniperda (Coleoptera: Curculionidae) in the Forest-Steppe Zone in Eastern Ukraine. European Journal of Entomology. 2014;111:561–5.
Liu DG, Bohne MJ, Lee JC, Flint ML, Penrose RL, Seybold SJ. New Introduction in California : The Redhaired Pine Bark Beetle. Hylurgus ligniperda Fabricius: color illustrations ma-USDA. 2007;1:3.
Ciesla WM. Recent Introductions of Forest Insects and Their Effects: A Global Overview. Plant Protection Bulletin. 1993;41:3–13.
David LM, Robert IG, Dolly L, Cecilia R, Sandra I, Rodrigo A. The Introduced Bark Beetles Hylurgus ligniperda and Hylastes ater (Coleoptera: Scolytidae) in Chile: Seasonal Flight and Effect of Pinus Radiata Log Placement on Colonization. Can J For Res. 2007;37:156–69.
Li C, Wang B, Ji Y, Huang L, Wang X, Zhao W, Wang Y, Wang H, Yao Y. Mitochondrial genome provides species-specific targets for the rapid detection of early invasive populations of Hylurgus ligniperda in China. BMC Genomics. 2024;25(1):90.
Kim S, Harrington TC, Lee JC, Seybold SJ. Leptographium Tereforme Sp Nov and Other Ophiostomatales Isolated from the Root-Feeding Bark Beetle Hylurgus ligniperda in California. Mycologia. 2011;103:152–63.
Gomez DF, Skelton J, De María M, Hulcr J. Influence of Temperature and Precipitation Anomaly on the Seasonal Emergence of Invasive Bark Beetles in Subtropical South America. Neotrop Entomol. 2020;49:347–52.
Tribe GD. Phenology of Pinus Radiata Log Colonization by the Red-Haired Pine Bark Beetle Hylurgus ligniperda (Fabricius) (Coleoptera: Scolytidae) in the South-Western Cape Province. J Entomol Soc South Afr. 1991;54:1–7.
David L Mausel, Robert I Gara, Dolly Lanfranco, Cecilia Ruiz, , Sandra Ide, and , Rodrigo Azat. The Introduced Bark Beetles Hylurgus ligniperda and Hylastes ater (Coleoptera: Scolytidae) in Chile: Seasonal Flight and Effect of Pinus Radiata Log Placement on Colonization. Canadian Journal of Forest Research. 2007;3:156–69.
Ayres MP, Lombardero MJ. Assessing the Consequences of Global Change for Forest Disturbance from Herbivores and Pathogens. Sci Total Environ. 2000;15:263–86.
Lanfranco D. An Analysis of Health Risk Reduction in Chilean Primary Forest Products for Export. Forestry. 2004;77:193–203.
Yang Y, Jiang HB, Liang CH, Ma YP, Dou W, Wang JJ. Chromosome-level genome assembly reveals potential epigenetic mechanisms of the thermal tolerance in the oriental fruit fly, Bactrocera dorsalis. Int J Biol Macromol. 2023;225:430–41.
King R, Buer B, Davies TGE, Ganko E, Guest M, Hassani-Pak K, et al. The complete genome assemblies of 19 insect pests of worldwide importance to agriculture. Pestic Biochem Physiol. 2023;191:105339.
Powell D, Groβe-Wilde E, Krokene P, Roy A, Chakraborty A, Löfstedt C, et al. A highly-contiguous genome assembly of the Eurasian spruce bark beetle, Ips typographus, provides insight into a major forest pest. Commun Biol. 2021;4(1):1059.
Keeling CI, Yuen MM, Liao NY, Docking TR, Chan SK, Taylor GA, et al. Draft Genome of the Mountain Pine Beetle, Dendroctonus Ponderosae Hopkins, a Major Forest Pest. Genome Biol. 2013;14:R27.
Liu Z, Xing L, Huang W, Liu B, Wan F, Raffa KF, et al. Chromosome-level genome assembly and population genomic analyses provide insights into adaptive evolution of the red turpentine beetle, Dendroctonus valens. BMC Biol. 2022;20(1):190.
Navarro-Escalante L, Hernandez-Hernandez EM, Nuñez J, Acevedo FE, Berrio A, Constantino LM, et al. A coffee berry borer (Hypothenemus hampei) genome assembly reveals a reduced chemosensory receptor gene repertoire and male-specific genome sequences. Sci Rep. 2021;11(1):4900.
Wang Z, Liu Y, Wang H, Roy A, Liu H, Han F, et al. Genome and transcriptome of Ips nitidus provide insights into high-altitude hypoxia adaptation and symbiosis. iScience. 2023;26(10):2589–0042.
Lifton RP, Goldberg ML, Karp RW, Hogness DS. The Organization of the Histone Genes in Drosophila Melanogaster: Functional and Evolutionary Implications. Cold Spring Harb Symp Quant Biol. 1978;42:1047–51.
Peleg S, Feller S, Forne I, Schiller E, Sévin DC, Schauer T, et al. Life Span Extension by Targeting a Link between Metabolism and Histone Acetylation in Drosophila. EMBO Rep. 2016;17:455–69.
Mixed CF, Oxidases F, Polyphagy H. The Devil’s Advocate Position. Ecological Entomology. 1984;9:29–34.
Bergé J, Feyereisen R, Amichot M. Cytochrome P450 Monooxygenases and Insecticide Resistance in Insects. Philos Trans R Soc Lond B Biol Sci. 1998;353:1701–5.
Debabrata P, Dunphy WG. Xe-P9, a Xenopus Suc1/Cks Protein, Is Essential for the Cdc2-Dependent Phosphorylation of the Anaphase- Promoting Complex at Mitosis. Genes Dev. 1998;12:2549–59.
Lee S, Lee R, Fraser A, Kamath R, Ahringer J, Ruvkun J. A Systematic RNAi Screen Identifies a Critical Role for Mitochondria in C. Elegans Longevity, Nature Genetics. 2003;33:40–8.
Li H, Durbin R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics. 2009;25:1754–60.
Weng YM, Francoeur CB, Currie CR, Kavanaugh DH, Schoville SD. A High-Quality Carabid Genome Assembly Provides Insights into Beetle Genome Evolution and Cold Adaptation. Mol Ecol Resour. 2021;21:2145–65.
Gagnaire PA, Pavey SA, Normandeau E, Bernatchez L. The genetic architecture of reproductive isolation during speciation-withgene-flow in lake whitefish species pairs assessed by RAD-sequencing. Evolution. 2013;67:2483–97.
Gao Q, Xiong Z, Larsen RS, Zhou L, Zhao J, Ding G, et al. High-Quality Chromosome-Level Genome Assembly and Full-Length Transcriptome Analysis of the Pharaoh Ant Monomorium Pharaonis. GigaScience. 2020;9:giaa143.
Navarro-Escalante L, Hernandez-Hernandez EM, Nuñez J, Acevedo FE, Berrio A, Constantino LM, et al. A Coffee Berry Borer (Hypothenemus hampei) Genome Assembly Reveals a Reduced Chemosensory Receptor Gene Repertoire and Male-Specific Genome Sequences. Sci Rep. 2021;11:4900.
Verhulst EC, van de Zande L. Insect sex determination: a cascade of mechanisms. Sex Dev. 2014;8(1–3):5–6.
Dafa’alla T, Fu G, Alphey L. Use of a regulatory mechanism of sex determination in pest insect control. J Genet. 2010;89:301–5.
Kawasaki Y, Schuler H, Stauffer C, Lakatos F, Kajimura H. Wolbachia endosymbionts in haplodiploid and diploid scolytine beetles (Coleoptera: Curculionidae: Scolytinae). Environ Microbiol Rep. 2016;8(5):680–8.
Keeling CI, Yuen MM, Liao NY, Docking TR, Chan SK, Taylor GA, et al. Draft genome of the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major forest pest. Genome Biol. 2013;14(3):R27.
Weinert BT, Timiras PS. Invited Review: Theories of Aging. J Appl Physiol. 2003;95:1706–16.
Hoffmann A, Sørensen JG, Loeschcke V. Adaptation of Drosophila to Temperature Extremes: Bringing Together Quantitative and Molecular Approaches. J Therm Biol. 2003;28:175–216.
How YF, Lee CY. Effects of Temperature and Humidity on the Survival and Water Loss of Cimex Hemipterus (Hemiptera: Cimicidae). J Med Entomol. 2010;47:987–95.
Owens ACS, Lewis SM. The Impact of Artificial Light at Night on Nocturnal Insects: A Review and Synthesis. Ecol Evol. 2018;8:11337–58.
Hussain M, Lin YW, Wang LD. Effect of Temperature on Longevity of Diaphorina Citri (Hemiptera: Liviidae) Studied by Microcalorimeter. J Therm Anal Calorim. 2017;127:1245–52.
Farmer KJ, Sohal RS. Effects of Ambient Temperature on Free Radical Generation, Antioxidant Defenses and Life Span in the Adult Housefly. Musca Domestica Experimental Gerontology. 1987;22:59–65.
Emana GD. Comparative Studies of the Influence of Relative Humidity and Temperature on the Longevity and Fecundity of the Parasitoid, Cotesia Flavipes. J Insect Sci. 2007;7:1–7.
Lu YH, Wu KM. Effect of Relative Humidity on Population Growth of Apolygus Lucorum (Heteroptera: Miridae). Appl Entomol Zool. 2011;46:421–7.
Scott JG, Wen Z. Cytochromes P450 of Insects: The Tip of the Iceberg. Pest Manag Sci. 2001;57:958–67.
Huber DP, Erickson ML, Leutenegger CM, Bohlmann J, Seybold SJ. Isolation and Extreme Sex-Specific Expression of Cytochrome P450 Genes in the Bark Beetle, Ips Paraconfusus, Following Feeding on the Phloem of Host Ponderosa Pine. Pinus Ponderosa Insect Molecular Biology. 2007;16:335–49.
Aw T, Schlauch K, Keeling CI, Young S, Bearfield JC, Blomquist GJ, Tittiger C. Functional Genomics of Mountain Pine Beetle (Dendroctonus Ponderosae) Midguts and Fat Bodies. BMC Genomics. 2010;11:1–12.
López MF, Cano-Ramírez C, Cesar-Ayala AK, Ruiz EA, Zúñiga G. Diversity and Expression of P450 Genes from Dendroctonus Valens LeConte (Curculionidae: Scolytinae) in Response to Different Kairomones. Insect Biochem Mol Biol. 2013;43:417–32.
Hýbl M, Mráz P, Šipoš J, Hoštičková I, Bohatá A, Čurn V, Kopec T. Polyphenols as Food Supplement Improved Food Consumption and Longevity of Honey Bees (Apis Mellifera) Intoxicated by Pesticide Thiacloprid. Insects. 2021;12:572.
Fabre JP, Carle P. Contribution à l’étude biologique d’Hylurgus Ligniperda F. (Coleoptera Scolytidae ) dans le Sud-est de la France. Annales des Sciences Forestières. 1975;32:55–71.
Hepat R, Kim Y. Transient Expression of a Viral Histone H4 Inhibits Expression of Cellular and Humoral Immune-Associated Genes in Tribolium Castaneum. Biochem Biophys Res Commun. 2011;415:279–83.
Amalia I, Aida FA, Cruz DL, Walker DW, Edgar BA. Changes in Neuronal CycD/Cdk4 Activity Affect Aging, Neurodegeneration, and Oxidative Stress. Aging Cell. 2015;14:896–906.
Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70.
Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications. 2020;11(1):1432.
Liu BH, Shi YJ, Yuan JY, Hu XS, Zhang H, Li N, et al. Estimation of Genomic Characteristics by Analyzing K-Mer Frequency in de Novo Genome Projects. Biology. 2013;35:62–7.
Sun HQ, Ding J. Mathieu Piednoël, Korbinian Schneeberger, FindGSE : Estimating Genome Size Variation within Human and Arabidopsis Using k -Mer Frequencies. Bioinformatics. 2018;34:550–7.
Neva C, Durand MS, Shamim IM, Suhas SPR, Huntley MH, Lander ES, Aiden EL. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–8.
Dudchenko O, Sanjit S,Arina D B, Sarah K O. Nyquist, et al. De Novo Assembly of the Aedes Aegypti Genome Using Hi-C Yields Chromosome-Length Scaffolds. Science. 2017;356:92–95.
Robinson JT, Turner D, Durand NC, Thorvaldsdóttir H, Mesirov JP, Aiden EL. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016;3:99–101.
Xu GC, Xu TJ, Zhu R, Zhang Y, Li SQ, Wang HW, Li JT. LR_Gapcloser: A Tiling Path-Based Gap Closer That Uses Long Reads to Complete Genome Assembly. GigaScience. 2019;8:giy157.
Jin JJ, Yu WB, Yang JB, Song Y, Claude W, Yi TS, Li DZ. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2010;21:1–31.
Hu J, Fan JP, Sun ZY, Liu SL. NextPolish: A Fast and Efficient Genome Polishing Tool for Long-Read Assembly. Bioinformatics. 2020;36:2253–5.
Pryszcz LP, Gabaldón T. Redundans: An Assembly Pipeline for Highly Heterozygous Genomes. Nucleic Acids Res. 2016;44: e113.
Li H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv. 2013: org 16.
Li H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Inanc Birol, Bioinformatics. 2018;34:3094–100.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15.
Simão FA, Waterhouse RM, Panagiotis L, Kriventseva EV, Zdobnov EM. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics. 2015;31:3210–2.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10(2):giab008.
Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, et al. Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined. Comprehensive Pipeline Genome Biology. 2019;20:275.
Lowe TM, Eddy SR. TRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997;25:955–64.
Kalvari I, Argasinska J, Quinones-Olvera N, Nawrocki EP, Rivas E, Eddy SR, et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Research. 2018;46(D1):D335–42.
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, Holt C, Sánchez Alvarado A, Yandell M. MAKER: An Easy-to-Use Annotation Pipeline Designed for Emerging Model Organism Genomes. Genome Res. 2008;18:188–96.
Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, et al. Automated Eukaryotic Gene Structure Annotation Using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:1–22.
Zhang RG, Li GY, Wang XL, Dainat J, Wang ZX, Ou S, Ma YP. TEsorter: An Accurate and Fast Method to Classify LTR-Retrotransposons in Plant Genomes. Horticulture Research. 2022;9:uhac017.
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P. Fast Genome-Wide Functional Annotation through Orthology Assignment by EggNOG-Mapper. Mol Biol Evol. 2017;34:2115–22.
Benjamin B, Xie C, Huson DH. Fast and Sensitive Protein Alignment Using DIAMOND. Nat Methods. 2015;12:59–60.
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics. 2014;30:1236–40.
Emms DM, Kelly S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019;20:1–14.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14(6):587–9.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol Biol Evol. 2015;32:268–74.
Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24:1586–91.
Hahn MW, De Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the Tempo and Mode of Gene Family Evolution from Comparative Genomic Data. Genome Res. 2005;15:1153–60.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X. MCScanX: A Toolkit for Detection and Evolutionary Analysis of Gene Synteny and Collinearity. Nucleic Acids Res. 2012;40:e49–e49.
Edgar RC. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004;32:1792–7.
Suyama M, Torrents D, Bork P. PAL2NAL: Robust Conversion of Protein Sequence Alignments into the Corresponding Codon Alignments. Nucleic Acids Res. 2006;34:W609–12.
Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genomics Proteomics Bioinformatics. 2006;4:259–63.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28(16):2184–5.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Yoav B, Yosef H. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc: Ser B (Methodol). 1995;57(1):289–300.
Wang H, Wen H, Li Y, Zhang K, Liu Y. Evaluation of potential reference genes for quantitative RT-PCR analysis in spotted sea bass (Lateolabrax maculatus) under normal and salinity stress conditions. PeerJ. 2018;19(6):e5631.
Wang WK, SONG JL, Yan J, Lu N, Yuan WD, Zhou ZF. Analysis of gene differential expressions between mycelia and fruiting bodies of Sanghuangporus sanghuang. Mycosystema. 2020;39(10):1874–1885.
Acknowledgements
We would like to thank Yantai Linfeng Tree Pest Control Co., Ltd. for their assistance in the sample collection process.
Funding
This work is supported by National Key Research & Development Program of China (2021YFD1400300).
Author information
Authors and Affiliations
Contributions
Zhiqian Chen collected the samples used in this paper. Quanzheng Yun, Jiaxing Li and Zhiqian Chen performed the genome sequencing, assembly, annotation and comparative genomics analysis. Zhiqian Chen wrote the manuscript. Jiaxing Li,,Ningning Fu, Youqing Luo and Lili Ren designed the works of the whole paper and helped to draft the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, Z., Ren, L., Li, J. et al. Chromosomal-level genome assembly of Hylurgus ligniperda: insights into host adaptation and environmental tolerance. BMC Genomics 25, 792 (2024). https://doi.org/10.1186/s12864-024-10711-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10711-6