
Abstract
Background
Phelan-McDermid syndrome (PMS) is a genetic condition characterized by intellectual disability, speech and language deficits, hypotonia, autism spectrum disorder, and epilepsy. PMS is caused by 22q13.33 deletions or mutations affecting SHANK3, which codes for a critical scaffolding protein in excitatory synapses. SHANK3 variants are also known to be associated with an increased risk for regression, as well as for psychiatric disorders, including bipolar disorder and catatonia. This study aimed to further describe these phenomena in PMS and to explore any relationship between psychiatric illness and regression after early childhood.
Methods
Thirty-eight people with PMS were recruited to this study through the Phelan-McDermid Syndrome Foundation based on caregiver report of distinct development of psychiatric symptoms. Caregivers completed a clinician-administered semi-structured interview focused on eliciting psychiatric symptomatology. Data from the PMS International Registry were used to confirm genetic diagnoses of participants and to provide a larger sample for comparison.
Results
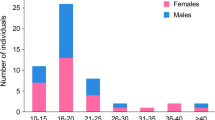
The mean age of the 38 participants was 24.7 years (range = 13 to 50; SD = 10.06). Females (31 of 38 cases; 82%) and sequence variants (15 of 38 cases; 39%) were over-represented in this sample, compared to base rates in the PMS International Registry. Onset of psychiatric symptoms occurred at a mean age of 15.4 years (range = 7 to 32), with presentations marked by prominent disturbances of mood. Enduring substantial loss of functional skills after onset of psychiatric changes was seen in 25 cases (66%). Symptomst indicative of catatonia occurred in 20 cases (53%). Triggers included infections, changes in hormonal status, and stressful life events.
Conclusions
This study confirms that individuals with PMS are at risk of developing severe neuropsychiatric illness in adolescence or early adulthood, including bipolar disorder, catatonia, and lasting regression of skills. These findings should increase the awareness of these phenotypes and lead to earlier diagnosis and the implementation of appropriate interventions. Our findings also highlight the importance of genetic testing in the work-up of individuals with intellectual disability and acute psychiatric illness or regression. Future research is needed to clarify the prevalence and nature of psychiatric disorders and regression among larger unbiased samples of individuals with PMS.
Similar content being viewed by others

Background
Phelan-McDermid syndrome (PMS) is a neurogenetic syndrome caused by haploinsufficiency of the SHANK3 gene due to a spectrum of anomalies in the terminal region of the long arm of chromosome 22, ranging from single-nucleotide variants to large deletions affecting multiple genes [1]. SHANK3 is a key structural protein in excitatory synapses, with several isoforms with different functions in development and in the synapse [2, 3]. Deletions or sequence variants of the SHANK3 gene are associated with the neuropsychiatric manifestations of the syndrome and required for the diagnosis of PMS [4,5,6,7]. Individuals with PMS often present with intellectual disability (ID), features of autism spectrum disorder (ASD), hypotonia, and severely delayed or absent speech [8,9,10]. Genotype-phenotype studies in individuals with terminal deletions have found that the severity of speech impairment and intellectual disability increases with increasing deletion size [8, 9]. In one recent study examining phenotypic manifestations among 17 individuals with sequence variants within SHANK3 [7], the majority developed single words, and 44% had phrase speech, in contrast to more severe impairment in individuals with deletions.
Epilepsy is reported in 17% to 70% of individuals with PMS [9, 11,12,13,14]. As individuals with PMS age, they appear to be at increased risk for bipolar disorder [15,16,17,18,19,20] and an associated risk of significant cognitive and behavioral regression [5, 6, 16, 18,19,20]. Indeed, SHANK3 variants have been implicated in the risk for severe neuropsychiatric disorders, including mood and psychotic disorders [15, 16, 19, 21, 22]. Gauthier et al. (2010) identified sequence variants in the SHANK3 gene in four individuals initially diagnosed with atypical, early-onset schizophrenia, and a history of borderline to mild intellectual disability [21]. Catatonia, a unique syndrome of motor and autonomic dysregulation associated with a variety of psychiatric and medical conditions, has also been described in PMS [7, 15, 22, 23]. Of note, catatonia in pediatric populations and in individuals with developmental disabilities often goes unrecognized [24] and poorly treated [25].
A few previous cross-sectional studies have used systematic methods to characterize the behavioral profile of individuals with PMS across the lifespan [19, 26,27,28]. However, the nature and course of psychiatric symptoms in PMS, and in particular their association with regression occurring much later than the early childhood regressions typical of ASD, have not been extensively documented. In this retrospective study, we collected developmental histories, behavioral profiles, and genetic findings of 38 adolescents and adults with PMS and psychiatric illness with the aim to (1) better characterize the psychiatric and developmental phenomena reported in PMS and (2) aid in early recognition and treatment optimization.
Methods
Participants and procedures
The study was approved by the Institutional Review Board of the PMS International Registry (PMSIR) (https://www.pmsf.org/registry/). Informed consent for participation in the Registry was obtained from the parents or legal guardians of participants, who also signed a release for inclusion in publication, and for integration of their interview data with their PMSIR data. Families were recruited by outreach through the PMS Foundation community Facebook page, and families were specifically invited to participate if the individual with PMS had experienced distinct psychiatric changes, such as mood episodes, psychosis, marked changes in sleep and energy, major loss of skills, sudden new intense obsessive-compulsive behaviors, or other neuropsychiatric difficulties, with or without regression. Families either contacted the researcher (TMK) directly in response to the Facebook message or responded after other families relayed the recruitment message on Facebook to parents with possible clinical concerns as described. In addition to the 37 English-speaking families, six families were referred through the PMS Spanish Association, and two enrolled and completed the study procedures.
Thirty-seven caregivers completed interviews on 39 participants and provided informed consent; one participant was excluded from the analysis since criteria were not met for a distinct psychiatric episode. Three families had responded to the invitation but did not complete interviews or consent after an initial contact established that they did not meet criteria for a distinct psychiatric episode. The final sample included 38 individuals from 36 families, ranging in age from 13 to 50 at the time of contact. The sample includes two sets of monozygotic twins with both twins enrolled. Caregivers interviewed were mothers in all but one case, in which the respondent was a sibling who was the legal guardian. Interviews were conducted in English (n = 34) or Spanish (n = 2), and respondents lived in the USA (n = 29), Australia (n = 4), Canada (n = 1), England (n = 1), Netherlands (n = 1), and Spain (n = 2).
Measures
A semi-structured interview entitled the Caregiver Interview for Psychiatric Illness in Persons with ID (see Additional file 1) was developed by TMK and conducted with caregivers by child and adolescent psychiatrists (TMK for English interviews and MPT for Spanish interviews). The interview includes questions designed to elicit descriptions of the participant’s developmental history, major health challenges, the emergence and course of episodes of psychiatric illness or regression, response to pharmacologic treatment, and current level of functioning. Interviews typically lasted 90 min. All final notes were reviewed and approved by the families before inclusion in the study.
Genetic reports were obtained for all participants and reviewed by the geneticist (CB) curating results for the PMSIR. PMSIR data were used to confirm genetic results for study participants and for comparisons of prevalence of genetic variants, age, and gender. However, only 21 of 38 participants in this study had completed the PMSIR clinical and developmental questionnaires, limiting direct comparisons of clinical data between study participants and other Registry members. Nine of 38 participants were not enrolled in the Registry prior to study participation.
Comparisons between functional status and developmental milestones in study participants vs Registry data included parent report of whether key developmental skills were ever achieved on the Caregiver Interview (for the study sample) and on the Registry’s Developmental Questionnaire for a group of respondents of similar age, with Registry data excluding the participants who were enrolled in the current study.
Assessment of behavioral symptoms and labeling of psychiatric diagnoses in the context of neurodevelopmental disorders is complicated by cognitive and communicative limitations, atypical presenting features, and premorbid characteristics (e.g., echolalia, repetitive behaviors). The Diagnostic and Statistical Manual for Mental Disorders, 5th edition [29] does not comprehensively include modifications of all diagnostic criteria for differential manifestations that may present in the context of intellectual disability, and there is a dearth of psychometrically rigorous instruments validated for psychiatric evaluation in this population. Therefore, we used the Diagnostic Manual - Intellectual Disability, Second Edition (DM-ID-2) [30] to classify psychiatric episodes. The DM-ID-2 is based on the DSM, adapted to include caregiver observations of behavior and to reduce the number of symptoms required to make diagnoses where persons with intellectual disability may not be able to report experiences [31]. All cases were reviewed by a child and adolescent psychiatrist (TMK) using a checklist of DM-ID-2 criteria for each of these disorders. The symptoms reported by caregivers during acute episodes met criteria for major depressive episode, manic episode, obsessive-compulsive disorder, generalized anxiety disorder, brief psychotic disorder, schizoaffective disorder, and catatonia associated with another mental disorder (see Additional file 2). In addition, two child and adolescent psychiatrists (MPT, AK) randomly reviewed and classified eight cases to assure accuracy of diagnostic classification. Agreement on the presence of a mood disorder was 100%, agreement on whether the first mood episode was depressive or manic was 88%, and agreement on the presence of catatonia was 88%.
The word “regression” is often used to describe a wide variety of states, including transient loss of skills during psychiatric episodes with and without catatonia. For clarity, “regression” was defined in this study as a prolonged loss of previously acquired skills that either (a) began when the individual was psychiatrically well or (b) began during a psychiatric episode, with loss of skills persisting for at least 6 months beyond the resolution of the psychiatric episode.
Results
Descriptive statistics and comparisons to participants in the PMSIR are reported in Table 1. Summaries of case histories and genetic findings are reported in Table 2. There was a strong female predominance in this sample (31 females, 7 males) whereas PMS typically affects males and females in equal proportion. There were 23 individuals with terminal deletions, with a mean size of 1.63 Mb (range 160 kb–6.41 Mb). Five deletions were secondary to a chromosomal rearrangement: a ring chromosome 22 in three individuals and an unbalanced translocation in two. Twelve individuals with terminal deletions had not had a karyotype, so the possibility of a ring chromosome 22 cannot be excluded. Fifteen individuals had pathogenic sequence variants in SHANK3 (12 frameshift variants and 3 nonsense variants).
As compared with the 509 PMSIR registrants not in this study who had genetic data available, sequence variants in SHANK3 were present at higher rates in our sample (39% [15/38] vs 6% [32/509]; Fisher’s exact test, p = 4 × 10− 8), and conversely, terminal deletions were less frequent (61% [23/38] vs 92% [467/509]; Fisher’s exact test, p = 8 × 10− 7). Comparisons with 130 PMSIR registrants of similar age with questionnaire data available are also reported in Table 1. In addition, deletions tended to be smaller in the participants of this study than in the PMSIR (mean size 1.634 Mb vs 3.633 Mb; unpaired two-tailed Student’s t test, p = 0.0019). In particular, individuals with small deletions, defined as those including at most the four distal genes, ARSA, SHANK3, ACR, and RABL2B, are over-represented among study participants compared to the PMSIR (57% [12/21 deletions with size information] vs 19% [72 /387]; Fisher’s exact test, p = 1.6 × 10− 4). (Heterozygous loss of ARSA, ACR, and RABL2B does not contribute to the PMS phenotype, so deletions including these genes are equivalent to deletions involving only SHANK3.)
Developmental history
Although documentation of the severity of ID was not available, reported levels of adaptive functioning suggest that mild to moderate ID was common in this sample prior to the onset of psychiatric symptoms and regression and that these individuals were less impaired prior to their psychiatric illness as compared to the PMSIR reference sample (Table 1). Prior to the onset of psychiatric illness, study participants were significantly more likely than participants in the PMSIR sample to ever have walked independently, achieved toilet training, verbal expression with at least phrase speech, and independence with dressing.
Psychiatric illness
Psychiatric difficulties began between ages 10 and 18 in the majority of cases (28/38; 74%). Psychiatric symptoms emerged between the ages of 7 and 10 years in 5/38 cases (13%), while 5/38 cases (13%) developed psychiatric symptoms between ages 21 and 32. Using the DM-ID-2, first episodes met criteria for a manic episode in 17/38 cases (45%) and for a depressive episode in 14/38 cases (37%). First episodes in the remaining cases included six (16%) with a mixture of mood and anxiety symptoms, and one case of disorganized, bizarre behavior that suggested a brief psychotic episode.
Most participants (27/38; 71%) had experienced a mood episode in the year before the study. The aftermath of acute episodes was associated with higher baseline levels of anxiety and irritability and with regression. Subsequent psychiatric episodes were superimposed and caregivers clearly distinguished episodes from baseline states. First episodes ranged from 8 months to 35 years before study participation. Reported episode length varied from periods of days to months. Most cases (34/38; 89%) had multiple distinct episodes consistent with a mood disorder, including 12 of 38 (32%) with two to four distinct episodes, 6 of 38 (16%) with five to nine episodes, and 16 of 38 (42%) with more than ten episodes (see Table 2 for details). Few cases (4/38; 11%) reported only a single psychiatric episode; all four of these were within the 3 years prior to study participation.
Twenty-nine of 38 cases (76%) reported a manic episode at some point in their illness; 24 of 38 (63%) reported both manic and depressive episodes. Nineteen of 38 cases (50%) were reported to have had psychotic symptoms during an acute mood episode. One participant presented with both depressive and manic episodes and with psychosis in between mood episodes, suggesting schizoaffective disorder.
New anxiety symptoms accompanied mood episodes in the majority of participants (26/38; 68%). Six participants (16%) met criteria for obsessive-compulsive disorder at some point in the course of their illness. Four participants (11%) met criteria for generalized anxiety disorder. Only two participants (5%) had first episodes that were predominantly anxious.
Symptoms consistent with a diagnosis of catatonia associated with another mental disorder were reported in 20 of 38 cases (53%). Seven of the 20 (35%) were on neuroleptics at the time of emergence of catatonic symptoms. Cases with sequence variants in SHANK3 met diagnostic criteria for catatonia significantly more often (12/15; 80%) than cases with terminal deletions (8/23; 35%) (Fisher’s exact test, p = 0.009).
Regression
The interview explored both abilities lost and the degree of return of skills over time. Caregivers were asked which skills were lost, at what age, and which, if any, skills were regained. Seven of the participants (18%) had experienced regressions in early childhood affecting communication, social interaction, and/or imaginative play (five by age two, one at age four, one at age seven), but we do not elaborate on those early regressions in the current report. Analysis focused on the 25 of 38 cases (66%) who had regressions that began within the 3 years after the onset of psychiatric episodes. Most of these regressions (21/25; 84%) began within a year after the onset of psychiatric episodes and involved multiple domains of function. Descriptions of the areas of skill lost and recovery are summarized in Table 2, which includes developmental status in terms of speech, toilet training, ability to dress and wash self, academic skills, and vocational activities when present. Caregiver reports of recovery of skills ranged from continuing loss of further skills to complete return to baseline function before the onset of psychiatric symptoms. Overall, more than half of the participants who regressed in the 3 years after the onset of psychiatric illness reported minimal recovery (14/25; 56%).
Antecedents to the onset of psychiatric illness and regression were explored in the interview. Menstrual cycling was reported to play a triggering role in 11 of 31 (35%) of the females. Acute infections were suspected triggers in 11 of 38 cases (29%). Psychosocial stressors were suspected triggers in 10 of 38 cases (26%). Several participants reported differing antecedent stressors for different episodes.
Co-morbid conditions
Only a quarter (9/38; 24%) of study participants was known to have PMS before the onset of psychiatric symptoms and associated regression. Sixteen cases (42%) had diagnoses of ASD before the onset of psychiatric symptoms and regression; five more received an ASD diagnosis after the onset of psychiatric illness and regression.
Fifteen of 38 cases (40%) reported one or more afebrile seizure. Eight of those 15 cases (53%) reported a seizure in the 2 years preceding the interview. Fifteen of 38 cases (40%) had weight loss of 10 to 25 kg accompanying psychiatric episodes. Chronic constipation, intermittent urinary incontinence, and episodes of acute urinary retention were prevalent in this sample, particularly as subjects aged. Chronic constipation was noted in 32 cases (84%), with episodes of acute urinary retention reported in 18 (47%). The discomfort associated with these conditions may lead to non-specific behaviors such as agitation, aggression, and self-injury in people with ID; these chronic symptoms were not coded as mood episodes. A small subset of subjects (4/38; 11%) had an immune-related disorder. Two participants (5%) were diagnosed with Hashimoto’s thyroiditis, one of whom was diagnosed with Hashimoto’s encephalopathy, and two participants (5%) had immunoglobulin deficiency, including one with common variable immunodeficiency.
Therapeutic interventions
At the time of interview, 32 of 38 participants (84%) were receiving one or more psychiatric medications, with 14 of 38 (37%) receiving four or more psychiatric medications. More than half (21/38; 55%) took anticonvulsants; all of these had mood symptoms, while 12 also had a history of seizures. Over half (55%) were receiving neuroleptics. Benzodiazepines were used either regularly or on an as-needed basis in 16 of 38 cases (42%). Alpha agonists, beta-blockers, antihistamines, and trazodone were prescribed for sleep or aggression. Smaller numbers of participants were receiving selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants, buspirone, lithium, cannabinoid oil, or n-acetylcysteine.
Full details of dosages and adjustments to treatment regimens in response to changing psychiatric status were inconsistently available and are therefore not reported. However, many parents kept extensive notes, and their reports of responses to medication trials suggest two patterns worth noting: (1) participants were reported to be sensitive to side effects from neuroleptics, with adverse events including extrapyramidal symptoms, worsening aggression and/or onset of catatonic symptoms reported in 14 of 30 cases (47%), while (2) seven of nine participants (78%) treated with SSRIs for depressive symptoms developed agitation, aggression, or other manic-like symptoms within weeks of the start of treatment.
Three individuals were treated with electroconvulsive therapy (ECT) for catatonia with reported significant benefit; two continue to require maintenance ECT (duration of treatment 18 months in one, > 8 years in the other) while the third discontinued after initial response due to agitation. One participant with recurrent catatonia on maintenance ECT was reported to have also benefited from intravenous immunoglobulin (IVIG) with improved cognition and motivation, but developed aseptic meningitis and a rash after the fifth treatment, leading to discontinuation of IVIG.
Nine participants (26%) were reported to have received immunomodulatory treatments for diagnoses of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) syndrome or of autoimmune encephalopathy. Six were diagnosed with PANDAS syndrome on the basis of clinical symptoms and positive strep cultures or elevated titers. One participant was diagnosed with Hashimoto’s encephalopathy, and two with seronegative autoimmune encephalitis. Treatments included (1) antibiotics, (2) IVIG, and/or (3) systemic anti-inflammatories such as nonsteroidal anti-inflammatory drugs, steroids, or rituximab.
In four of six cases diagnosed with PANDAS syndrome, parents reported that antibiotic treatment was effective in reducing or eliminating acute symptoms. The remaining two improved slowly; one also received IVIG and the other received lorazepam; these parents were not confident that antibiotics had played a role in recovery. Treatment for PANDAS symptoms did not appear to prevent future mood episodes or regression, as all but one of these cases went on to have further psychiatric episodes and some had later regressions. The one participant diagnosed with Hashimoto’s encephalopathy continues to receive regular infusions of IVIG with steroids, as well as rituximab. The two participants with diagnoses of seronegative autoimmune encephalopathy receive monthly IVIG.
Eight females were on hormonal therapy to eliminate menstrual cycles and reduce associated mood symptoms. Non-pharmacological treatments varied widely by geographic location and age, with those still in school more likely to be receiving structured behavioral interventions. Data on past and current behavioral programs and therapies were not included in the interview.
Discussion
This case series has characterized a subset of individuals with PMS who have episodic, severe neuropsychiatric illness, frequently beginning in adolescence or early adulthood, with major impacts on functional status. The symptoms and course of illness often closely resembled bipolar illness, with psychotic features almost exclusively present in the context of mood episodes. None of the participants in our sample presented with a primary psychotic disorder, such as schizophrenia. However, one participant presented with distinct psychotic and affective episodes characteristic of schizoaffective disorder. Using the DM-ID-2 criteria [30], all but one of the participants had either a mood disorder or an anxiety disorder, with multiple discrete periods of illness in all but four.
Several triggers were often reported as temporal antecedents to the onset of psychiatric changes. Biological triggers included infections and changes in hormonal status, while environmental factors included stressful life events. Similar patterns have been observed in other, more common neurogenetic syndromes, including Down syndrome [32], Williams syndrome [33], and 22q11.2 deletion syndrome [34].
It is critical to emphasize the high incidence of catatonic symptoms in this subset of individuals with PMS, as catatonia often goes unrecognized or undertreated in individuals with developmental disabilities [25, 35]. Recognition and treatment of both stuporous and hypermotoric catatonia is crucial, as symptoms can lead to life-threatening complications. Other genetic conditions involving copy number variants, such as 22q11.2 deletions [36], have also been associated with psychiatric symptoms and with catatonia. Similar to their use in treating catatonia in other conditions, benzodiazepines and ECT were used in this cohort with reported tolerability and effectiveness.
Regression has long been recognized in neurodevelopmental disorders such as ASD [37], but the triggers and mechanisms are not well understood, and the literature focuses largely on early childhood regression, especially as it relates to Rett syndrome [37,38,39,40]. Significant cognitive and behavioral regression has been documented in PMS [5, 6, 9, 15, 16, 18, 41]. Verhoeven et al. [20] have recently published on the course of illness of 24 adolescents and adults with PMS referred for evaluation and treatment of “challenging behaviors and unstable mood.” In that sample, there were five individuals with periodic catatonic symptoms, and four other individuals who developed progressive loss of skills in their third or fourth decade.
Two thirds (66%) of our sample described lasting regressions that began within 3 years of the onset of psychiatric illness. These regressions in communication, self-care, and motor functions left a subset of previously more capable individuals largely non-verbal, incontinent, and unable to dress or feed themselves. Many of these participants continue to experience episodic psychiatric illness in addition to developmental decline. Interestingly, a similar combination of rapid onset psychiatric disturbance with marked regression and catatonia has been described in Down syndrome [42, 43] and subsequently labeled “Down syndrome disintegrative disorder.” A recent case series [44] documented a role for immunotherapy in restoring function and stability in these individuals, although it is also recognized that cognitive decline in Down syndrome relates to amyloid precursor protein gene triplication [45,46,47]. Regression in PMS may be more common than in Down syndrome, and its potentially devastating impact warrants ongoing study of the natural history, mechanism, and targets for intervention.
Given the methodological limitations and sample selection bias in this study, we are unable to draw conclusions about the prevalence of psychiatric illness and/or regression among people with PMS. This study was also limited by the lack of standardized or validated tools to measure neuropsychiatric symptoms in severely developmentally disabled children and adults. However, within this self-referred subset of individuals with PMS whose caregivers identified psychiatric illness, we observed a distinct pattern of affective episodes, with onset typically in puberty, a strikingly high incidence of catatonic symptoms, and an association with substantial regression in the second through fourth decades of life. Of note, a recent literature review analyzed prior reports of 56 individuals with PMS with accompanying neuropsychiatric symptoms, and concluded that “clinicians and caregivers need to be vigilant for loss of skills and neuropsychiatric changes in adolescents and adults with PMS, including the development of bipolar disorder and catatonia” [48].
Individuals with PMS who present with new psychiatric symptoms require careful monitoring and cautious early intervention. Response to traditional psychiatric interventions is variable, and the role for immunomodulatory treatments and ECT in specific subsets of patients should be explored in future studies. The pattern of rapid and severe deterioration described in more than half of this group calls for further investigation to identify triggers and the underlying biology and to delineate possible strategies for prevention and timely intervention. While the current study does not allow for specific treatment recommendations, there is an urgent need for clinical trials to assess the efficacy of existing psychopharmacological treatments for psychiatric symptoms in PMS, in addition to developing novel, targeted therapeutics.
The preponderance of females in this sample (> 4:1) is notable and calls for replication. Sequence variants in SHANK3 were also six times more common in this sample than in the PMSIR. These findings raise questions about whether psychiatric problems and regression disproportionately affect females and/or individuals with SHANK3 sequence variants, in contrast to those with deletions. In addition, prior to the onset of psychiatric illness, most of the participants described herein were less severely impaired in their adaptive functioning than is reported in most studies of PMS. This finding is likely related to the high proportion of individuals with SHANK3 variants and small deletions in this sample.
Further studies should attempt to clarify if psychiatric difficulties are actually more common in individuals who are more verbal and social and who have more intact adaptive functioning at baseline, or, whether this finding is the result of selection bias in our sample, as the majority of participants (76%) were only diagnosed with PMS after the onset of psychiatric symptoms.
Finally, our sample underscores the importance of genetic testing as part of the medical work-up for individuals with this presentation. Chromosomal microarray is recommended for the evaluation of all children with global developmental delay or intellectual disability [49]. In the setting of superimposed psychiatric illness or regression, chromosomal microarray should be considered as first tier testing, followed by sequencing of SHANK3 if the microarray is unrevealing.
Conclusion
This study confirms that individuals with PMS are at risk of developing severe neuropsychiatric illness in adolescence or early adulthood. In most cases, the symptoms appear consistent with the expression of bipolar disorder in individuals with intellectual disability, with catatonia noted as a common co-occurring condition. Triggers may include infections, changes in hormonal status, and stressful life events. Significant cognitive and behavioral regression beyond a baseline level of disability has been previously reported in PMS and accompanied the onset of psychiatric illness in a majority of the cases reported here. Our results must be interpreted with caution given the potential selection bias in recruitment, but it is clear that people with PMS may show a rapid and severe deterioration that requires careful monitoring and intervention. Our findings also highlight the relevance of genetic testing in the work-up of individuals with intellectual disability and acute psychiatric illness or regression. Future research is needed to clarify the prevalence and nature of psychiatric symptoms and regression among larger unbiased samples of individuals with PMS and to delineate any shared mechanisms with other neurodevelopmental disorders presenting with psychiatric illness and/or regression in adolescence or early adulthood. Identification of early clinical and biological markers would contribute to our understanding of the underlying neurobiology of these disorders and potentially aid in monitoring, early intervention, or prevention.
Abbreviations
- ASD:
-
Autism spectrum disorder
- DM-ID-2:
-
Diagnostic Manual - Intellectual Disability, Second Edition
- ECT:
-
Electroconvulsive therapy
- ID:
-
Intellectual disability
- IVIG:
-
Intravenous immunoglobulin
- PANDAS:
-
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections
- PMS:
-
Phelan-McDermid syndrome
- PMSIR:
-
Phelan-McDermid Syndrome International Registry
References
Phelan K, Rogers RC, Boccuto L. Phelan-McDermid syndrome. In: Adam M, Ardinger H, Pagon R, Wallace S, Bean L, Stephens K, editors. Gene Reviews. Seattle: University of Washington; 1993.
Wang X, Xu Q, Bey AL, Lee Y, Jiang YH. Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol Autism. 2014;5:30.
Costales J, Kolevzon A. The therapeutic potential of insulin-like growth Factor-1 in central nervous system disorders. Neurosci Biobehav Rev. 2016;63:207–22. https://doi.org/10.1016/j.neubiorev.2016.01.001.
Wilson H, Wong A, Shaw S, Tse W, Stapleton G, Phelan MC, et al. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–84.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39(1):25–7.
Bonaglia MC, Giorda R, Beri S, de Agostini C, Novara F, Fichera M, et al. Molecular mechanisms generating and stabilizing terminal 22q13 deletions in 44 subjects with phelan/mcdermid syndrome. PLoS Genet. 2011;7(7):e1002173.
De Rubeis S, Siper PM, Durkin A, Weissman J, Muratet F, Halpern D, et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol Autism. 2018;9(1):1–20.
Sarasua SM, Boccuto L, Sharp JL, Dwivedi A, Chen CF, Rollins JD, et al. Clinical and genomic evaluation of 201 patients with Phelan-McDermid syndrome. Hum Genet. 2014;133(7):847–59. [cited 2015 Jan 12]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/24481935
Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol Autism. 2013;4(1):18. [cited 2015 Jan 12]; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3707861&tool=pmcentrez&rendertype=abstract
Kolevzon A, Angarita B, Bush L, Wang AT, Frank Y, Yang A, et al. Phelan-McDermid syndrome : a review of the literature and practice parameters for medical assessment and monitoring. J Neurodev Disord. 2014;6:39.
Khan O, Zhou X, Leon J, Kessler R, Gaughan T, D’Souza P, et al. Prospective longitudinal overnight video-EEG evaluation in Phelan-McDermid Syndrome. Epilepsy Behav. 2018;80:312–20.
Jeffries AR, Curran S, Elmslie F, Sharma A, Wenger S, Hummel M, et al. Molecular and phenotypic characterization of ring chromosome 22. Am J Med Genet A. 2005;137(2):139–47. [cited 2015 Jan 12]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/16059935
Dhar SU, Del Gaudio D, German JR, Peters SU, Ou Z, Bader PI, et al. 22q13.3 Deletion syndrome: clinical and molecular analysis using array CGH. Am J Med Genet Part A. 2010;152(3):573–81. [cited 2015 Jan 12]; Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3119894&tool=pmcentrez&rendertype=abstract
Holder JL, Quach MM. The spectrum of epilepsy and electroencephalographic abnormalities due to SHANK3 loss of function mutations. Epilepsia. 2016;57(10):1651–9.
Serret S, Thümmler S, Dor E, Vesperini S, Santos A, Askenazy F. Lithium as a rescue therapy for regression and catatonia features in two SHANK3 patients with autism spectrum disorder: case reports. BMC Psychiatry. 2015;15(1):107. Available from: http://www.biomedcentral.com/1471-244X/15/107%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/25947967%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC4428105
Denayer A, Van Esch H, De Ravel T, Frijns JP, Van Buggenhout G, Vogels A, et al. Neuropsychopathology in 7 patients with the 22q13 deletion syndrome: Presence of bipolar disorder and progressive loss of skills. Mol Syndromol. 2012;3(1):14–20. [cited 2015 Jan 12]. https://doi.org/10.1159/000339119.
Verhoeven WMA, Egger JIM, Cohen-Snuijf R, Kant SG, de Leeuw N. Phelan-McDermid syndrome: clinical report of a 70-year-old woman. Am J Med Genet A. 2013;161A(1):158–61. [cited 2015 Jan 12]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/23166010
Vucurovic K, Landais E, Delahaigue C, Eutrope J, Schneider A, Leroy C, et al. Bipolar affective disorder and early dementia onset in a male patient with SHANK3 deletion. Eur J Med Genet. 2012;55(11):625–9. [cited 2015 Jan 12]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22922660
Egger JIM, Zwanenburg RJ, Van Ravenswaaij-Arts CMA, Kleefstra T, Verhoeven WMA. Neuropsychological phenotype and psychopathology in seven adult patients with Phelan-McDermid syndrome: implications for treatment strategy. Genes Brain Behav. 2016;15:395–404.
Verhoeven WMA, Egger JIM, de Leeuw N. A longitudinal perspective on the pharmacotherapy of 24 adult patients with Phelan McDermid syndrome. Eur J Med Genet. 2019:103751. https://doi.org/10.1016/j.ejmg.2019.103751.
Gauthier J, Champagne N, Lafrenière RG, Xiong L, Spiegelman D, Brustein E, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci. 2010;107:7863–8.
Breckpot J, Vercruyssen M, Weyts E, Vandevoort S, D’Haenens G, Van Buggenhout G, et al. Copy number variation analysis in adults with catatonia confirms haploinsufficiency of SHANK3 as a predisposing factor. Eur J Med Genet. 2016;59:436–43.
Jungová P, Čumová A, Kramarová V, Lisyová J, Ďurina P, Chandoga J, et al. Phelan-McDermid syndrome in adult patient with atypical bipolar psychosis repeatedly triggered by febrility. Neurocase. 2018;24(4):227–30. https://doi.org/10.1080/13554794.2018.1542007.
Benarous X, Consoli A, Raffin M, Bodeau N, Giannitelli M, Cohen D, et al. Validation of the Pediatric Catatonia Rating Scale (PCRS). Schizophr Res. 2016;176:378–86.
Dhossche DM, Wachtel LE. Catatonia is hidden in plain sight among different pediatric disorders: a review article. Pediatr Neurol. 2010;43:307–15.
Soorya L, Leon J, Trelles MP, Thurm A. Framework for assessing individuals with rare genetic disorders associated with profound intellectual and multiple disabilities (PIMD): the example of Phelan McDermid syndrome. Clin Neuropsychol. 2018;32:1226–55.
Richards C, Powis L, Moss J, Stinton C, Nelson L, Oliver C. Prospective study of autism phenomenology and the behavioural phenotype of Phelan-McDermid syndrome: comparison to fragile X syndrome, Down syndrome and idiopathic autism spectrum disorder. J Neurodev Disord. 2017;9:37.
Zwanenburg RJ, Ruiter SAJ, Van Den Heuvel ER, Flapper BCT, Van Ravenswaaij-Arts CMA. Developmental phenotype in Phelan- McDermid (22q13.3 deletion) syndrome: a systematic and prospective study in 34 children. J Neurodev Disord. 2016;8:16.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). Arlington:. American Journal of Psychiatry. 2013. American Psychiatric Association. (2013). Diagnostic and statistical manual of mental disorders (5th ed.). Arlington, VA:
Fletcher RJ, National Association for the Dually Diagnosed., & American Psychiatric Association. DM-ID: Diagnostic Manual Intellectual Disability : a clinical guide for diagnosis of mental disorders in persons with intellectual disability. Kingston: National Association for the Dually Diagnosed; 2007.
Fletcher RJ, Barnhill J, McCarthy J, Strydom A. From DSM to DM-ID. J Ment Health Res Intellect Disabil. 2016;9:189–204.
Dykens EM, Shah B, Davis B, Baker C, Fife T, Fitzpatrick J. Psychiatric disorders in adolescents and young adults with Down syndrome and other intellectual disabilities. J Neurodev Disord. 2015;7:9.
Valdes F, Keary CJ, Mullett JE, Palumbo ML, Waxler JL, Pober BR, et al. Brief report: major depressive disorder with psychotic features in Williams syndrome: a case series. J Autism Dev Disord. 2018;48:947–52.
Schneider M, Debbané M, Bassett AS, Chow EWC, Fung WLA, Van Den Bree MBM, et al. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry. 2014;171:627–39.
Fink M, Taylor MA, Ghaziuddin N. Catatonia in autistic spectrum disorders: a medical treatment algorithm. Int Rev Neurobiol. 2006;72:233–44.
Butcher NJ, Boot E, Lang AE, Andrade D, Vorstman J, McDonald-McGinn D, et al. Neuropsychiatric expression and catatonia in 22q11.2 deletion syndrome: an overview and case series. Am J Med Genet Part A. 2018;176:2146–59.
Barger BD, Campbell JM, McDonough JD. Prevalence and onset of regression within autism spectrum disorders: a meta-analytic review. J Autism Dev Disord. 2013;43:817–28.
Thurm A, Powell EM, Neul JL, Wagner A, Zwaigenbaum L. Loss of skills and onset patterns in neurodevelopmental disorders: understanding the neurobiological mechanisms. Autism Res. 2018;11:212–22.
Gadow KD, Perlman G, Weber RJ. Parent-reported developmental regression in autism: epilepsy, IQ, schizophrenia Spectrum symptoms, and special education. J Autism Dev Disord. 2017;47(4):918–26.
Einspieler C, Marschik PB. Regression in Rett syndrome: developmental pathways to its onset. Neurosci Biobehav Rev. 2019;98:320–32.
Reierson G, Bernstein J, Froehlich-Santino W, Urban A, Purmann C, Berquist S, et al. Characterizing regression in Phelan McDermid syndrome (22q13 deletion syndrome). J Psychiatr Res. 2017;91:139–44.
Worley G, Crissman BG, Cadogan E, Milleson C, Adkins DW, Kishnani PS. Down syndrome disintegrative disorder: new-onset autistic regression, dementia, and insomnia in older children and adolescents with down syndrome. J Child Neurol. 2015;30:1147–52.
Ghaziuddin N, Nassiri A, Miles JH. Catatonia in Down syndrome; a treatable cause of regression. Neuropsychiatr Dis Treat. 2015;11:941.
Cardinale KM, Bocharnikov A, Hart SJ, Baker JA, Eckstein C, Jasien JM, et al. Immunotherapy in selected patients with Down syndrome disintegrative disorder. Dev Med Child Neurol. 2018;61:847–51.
Lao PJ, Handen BL, Betthauser TJ, Mihaila I, Hartley SL, Cohen AD, et al. Alzheimer-like pattern of hypometabolism emerges with elevated amyloid-β Burden in Down syndrome. J Alzheimers Dis. 2017;61:631–44.
Hartley SL, Handen BL, Devenny D, Mihaila I, Hardison R, Lao PJ, et al. Cognitive decline and brain amyloid-β accumulation across 3 years in adults with Down syndrome. Neurobiol Aging. 2017;58:68–76.
Startin CM, Ashton NJ, Hamburg S, Hithersay R, Wiseman FK, Mok KY, et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer’s disease. Alzheimers Res Ther. 2019;11(1):1–12.
Kolevzon, A., Delaby, E., Berry-Kravis, E. et al. Neuropsychiatric decompensation in adolescents and adults with Phelan-McDermid syndrome: a systematic review of the literature. Molecular Autism. 2019: https://doi.org/10.1186/s13229-019-029-3.
Moeschler JB, Shevell M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134:e903–18.
Acknowledgements
We thank the families for their many contributions and the Phelan-McDermid Syndrome Foundation for its support.
Funding
MPT receives research support from the National Institute of Health National Research Service Award Institutional Training Program (T32) and the Beatrice and Samuel A. Seaver Foundation. AT receives support from the Intramural Research Program of the National Institute of Mental Health (ZICMH002961). AK receives support from the National Institute of Neurological Disorders and Stroke (R01NS105845-01 and U54 NS092090-01) and from the Beatrice and Samuel A. Seaver Foundation.
Availability of data and materials
The datasets analyzed in the current study are available at the PMS International Registry to authorized users (https://www.pmsf.org/registry/).
Author information
Authors and Affiliations
Contributions
TK and MPT collected the data. BL and CB performed the analyses of the PMS International Registry data. CB reviewed all genetic reports and performed analyses of the relationship between genetic results and clinical findings. All authors were involved in drafting or revising the manuscript, have read and agreed to its content, and are accountable for all aspects of the accuracy and integrity of the manuscript, in accordance with ICMJE criteria.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Informed consent was obtained for participants, who were enrolled in the Phelan-McDermid Syndrome International Registry (https://www.pmsf.org/registry/).
Consent for publication
All participants signed a release for publication of results.
Competing interests
AK receives research support from AMO Pharma and consults to Ovid Therapeutics, Takeda, 5 AM Ventures, sema4, and LabCorp. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1.
Caregiver Interview for Psychiatric Illness in Persons with ID (CIPIPID)
Additional file 2.
Adapted Diagnostic Criteria, DM-ID-2
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kohlenberg, T.M., Trelles, M.P., McLarney, B. et al. Psychiatric illness and regression in individuals with Phelan-McDermid syndrome. J Neurodevelop Disord 12, 7 (2020). https://doi.org/10.1186/s11689-020-9309-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11689-020-9309-6