
Abstract
In a previous study, we reported a transient transformation system using repeated screening for hygromycin B (Hyg) resistance in the basidiomycete Ceriporiopsis subvermispora. In the present study, by combining this technique with CRISPR/Cas9, we demonstrated successful marker-free genome editing in Pleurotus ostreatus, which is one of the most economically important cultivated mushrooms as well as a model white-rot fungus. At first, transformant selection mediated by the transient expression of marker genes was demonstrated using a plasmid harboring the Hyg resistance gene (hph) in P. ostreatus. Then, genome editing of fcy1, which confers 5-fluorocytosine (5-FC) resistance to the host cell, was performed by the transient expression of Cas9, gRNA, and hph and strains with 5-FC resistance and Hyg sensitivity were isolated. Additionally, genome editing of fcy1 in these strains was confirmed by Sanger sequencing. To our knowledge, this is the first report of marker-free genome editing through the transient expression of Cas9, gRNA, and hph in agaricomycetes, which opens the door for repeated genome editing in these fungi.
Similar content being viewed by others

Introduction
Genetic transformation is a basic tool for the functional analyses of the genes of interest, as well as for molecular breeding. Generally, genetic transformations can be divided into stable and transient transformations [1, 2]. In stable transformation, the foreign DNA is mostly integrated into the host chromosome. In the case of transient transformation, the introduced DNA resides temporarily in the cell and is subsequently lost because chromosome integration does not occur. Both transformation methods have been used for genetic investigations in plants and animal cells. In filamentous fungi, stable transformation has been widely used to integrate genes at random or targeted sites on chromosomes. A substantial number of different selection markers are required for multiple gene integrations or disruptions, limiting such genetic manipulation. Transformation of Schizophyllum commune and Coprinopsis cinerea was first reported in basidiomycetes [3, 4]. Stable transformation has been developed for the genetic investigations of several edible mushrooms [5,6,7]. Recently, we reported the first transient transformation of the basidiomycete Ceriporiopsis (Gelatoporia) subvermispora [8]. Transient transformants carrying temporary hygromycin B (Hyg) resistance were obtained only when a functional promoter was used to express the coding sequence of the Hyg resistance gene (hph). This result demonstrated that the unstable Hyg-resistant colonies were not background colonies but were dependent on the transient expression of the introduced recombinant hph. If this system can be applied to other fungi, repetitive use of the selection marker for multiple transformations becomes possible, which opens the door for the multiple gene disruptions or insertions.
Clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated protein 9 (Cas9) has become the most popular genome-editing technology [9]. CRISPR/Cas9 was originally found in bacteria and archaea which functions as an adaptive immune system against invading nucleic acids. In this system, a 20-nt guide RNA (gRNA) recruits the Cas9 protein to the target site, and Cas9 cuts the polynucleotide linkages to make a double-strand break in the host genome. During the break repair process, a mutation, such as the deletion or insertion of a nucleotide sequence, may occur in the host genome. In the presence of donor DNA containing flanking sequence surrounding the double-strand break, a gene knock-in can occur through homologous recombination at the gRNA target site. CRISPR/Cas9 was successfully adapted for the agaricomycete C. cinerea in 2017 [10]. Recently, we developed the first CRISPR/Cas9 systems for edible mushrooms using P. ostreatus [11, 12].
White-rot fungi, including P. ostreatus, have a unique ability to depolymerize wood lignin. Generally, it is difficult to effectively remove lignin from woody biomass under mild conditions to obtain polysaccharides for biorefineries. Therefore, the molecular mechanism underlying lignin biodegradation by white-rot fungi is of interest to establish new methods for efficient and ecofriendly lignin removal from wood biomass [13]. Multiple genes encoding ‘ligninolytic enzymes’ have been predicted in the P. ostreatus genome [14]. In the case of extracellular peroxidase, three genes encoding versatile peroxidases (VPs) and six genes encoding manganese peroxidases (MnPs) were predicted in the P. ostreatus genome [15]. To date, there have been no reports directly demonstrating that any of these extracellular peroxidases are involved in lignin degradation by white-rot fungi. Salame et al. isolated single gene disruptants of mnp or vp using gene knock-out in a non-homologous end joining (NHEJ)-deficient strain, but these strains did not show a drastic decrease in their lignin degradation ability on wood sawdust medium [16,17,18]. These results suggest that the enzymes encoded by the disrupted genes are not essential for ligninolysis, but it is important to consider the redundancy of these genes and their effect on the phenotype of the single mnp or vp disruptants. Therefore, multiple gene disruptions are required to understand the physiological function of each ligninolytic enzyme.
To knock-out a gene on a chromosome, conventional gene targeting through homologous recombination-based replacement of a target site with a selectable marker gene is required. As available selection markers are limited, multiple gene targeting is very difficult in this fungus [19]. This limitation has hindered efforts to disrupt multiple genes encoding lignin-degrading enzymes. Recently, CRISPR/Cas9-directed genome editing was used to introduce mutations at targeted sites on the chromosome and to isolate disruptants of a gene of interest. However, in this case, a selection marker is still essential to isolate the mutant strains, and there is a limitation for repetitive genome-editing experiments owing to the shortage of available selection markers. If a practical method for temporal CRISPR/Cas9 and marker selection could be established, repetitive genome editing would become available, permitting multiple gene disruptions of ligninolytic enzyme genes to elucidate their physiological functions in wood degradation by P. ostreatus.
In this study, marker selection of the transient transformants dependent on the temporal expression of hph was established in P. ostreatus. Then, marker-free genome editing was performed by combining the transient expression of Cas9, gRNA, and hph.
Materials and methods
Strains, plasmids, culture conditions, and genetic techniques
Pleurotus ostreatus monokaryon strain PC9 (Spanish Type Culture Collection accession number CECT20311) was used for transformation in this study [20]. The 5-fluorocytocine (5-FC) sensitivity of this strain was confirmed in our previous study [21]. The strains used in this study are listed in Table 1. Yeast and malt extract with glucose (YMG) agar were used for routine culture [22]. The cultures were maintained at 28 °C in continuous darkness unless otherwise stated.
Transformations using the hygromycin B resistance gene (hph) were performed as described previously [16]. For the selection of transformants with hph, YMG supplemented with 100 μg/mL hygromycin B (YMG-Hyg) was used. YMG supplemented with 0.1% (w/v) 5-FC (YMG-5FC) was used to select for the fcy1 mutant.
The plasmids pPHT1 [23], pCcPef3-126 [24], and pCcPef3-126-fcy1sg2 [11] were constructed as previously described.
Rapid colony PCR and sequencing
Crude genomic DNA was prepared as described previously [25]. The PCRs described below were performed using PrimeSTAR GXL DNA polymerase (Takara Bio) or KOD FX neo DNA polymerase (TOYOBO, Osaka, Japan). The primers used for the colony PCR are listed in Table 2.
The cas9 insertion was confirmed by amplifying the 2232-bp fragment of the cas9 gene using PCR with primers Cas9check_F2 and Cas9check_R2. The hph insertion was confirmed by amplifying the 1012-bp fragment of the hph gene using PCR with primers hph_F and hph_R. Genome editing of fcy1 was detected by PCR with primers TB9 and TB10, and Sanger sequencing was performed against the resulting PCR product using the same primer sets (Fig. 3).
Results and discussion
Selection of transformants through transient expression of hph in P. ostreatus
Previously, transient expression of a selection marker was observed by introducing a plasmid harboring an hph expression cassette into C. subvermispora, which can be used for initial selection of transformants [8]. If transient hph expression can be used for the initial selection of transformants in P. ostreatus, marker-free genome editing will be possible in combination with the transient expression of Cas9 and gRNA. To determine whether transient hph expression could be used for initial selection of transformants in P. ostreatus, the pPHT1 plasmid harboring an hph expression cassette was introduced into wild-type P. ostreatus. A scheme of the transient expression of hph by introduction of pPHT1 is shown in Fig. 1a, b. In the first screening, transformants were selected on YMG-Hyg agar medium. From this plate, 46 viable strains were isolated and transferred to fresh YMG agar medium. After growth to reach the edge of the agar plate, transformants were passaged on YMG-Hyg agar medium to test for Hyg resistance. Three of the 46 strains showed loss of Hyg resistance. The presence of the hph gene in these strains was tested using colony PCR. The results showed that hph was amplified in Hyg-resistant strains, whereas no bands were detected in these three strains (Fig. 1c). These results indicate that temporal expression of hph occurred in P. ostreatus and could be applied to the initial selection of transformants. It is notable that transient hph expression can be used for initial screening of transformants in two taxonomically separated species, P. ostreatus and C. subvermispora, suggesting the possibility of a similar selection system in other basidiomycete fungi.

Transient hph expression in Pleurotus ostreatus. a Schematic diagram for transient hph expression. b Plasmid map of pPHT1. CctubB_pro or ter: promoter or terminator from tubulin gene in Coprinopsis cinerea, ColE1 ori: origin for Escherichia coli, KmR: kanamycin resistance gene, AmpR: ampicillin resistance gene, hph: hygromycin phosphotransferase. c PCR amplification of hph fragment using primer set hph_F and hph_R. Lane M, 1-kb ladder (Nippon gene); lane P, pPHT1 (positive control); lane W, parental strain PC9 (negative control); lanes 1–5, PHT1-1–5 strains. HygR: hygromycin B resistant, HygS: hygromycin B sensitive
Marker-free genome editing by transient expression of Cas9, gRNA, and hph
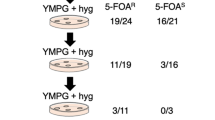
In the previous study, we have developed CRISPR/Cas9 system for P. ostreatus [26]. However, it has not indicated that if genome editing is available via the transient expression of Cas9 and gRNA. Additionally, it is unsure whether transient expression of selection marker can be used for initial selection of genome-edited strains. To examine gene knock-out by transient Cas9, gRNA, and hph expression, the fcy1 gene was selected as a target because loss of its function confers resistance to 5-FC in P. ostreatus [21]. This allowed us to identify mutants easily and efficiently by examining resistance/sensitivity to 5-FC after screening for temporal Hyg resistance during the initial screening. A schematic of the experimental design is shown in Fig. 2a. The plasmid pCcPef3-126-fcy1sg2, which was constructed for genome editing of fcy1 in a previous study [11], was introduced into P. ostreatus, and transformants were selected on YMG-Hyg with sucrose agar medium (Fig. 2b). The obtained Hyg-resistant transformants were subcultured on YMG-5FC agar medium and subjected to the following analysis. From these transformants, 130 5-FC-resistant transformants were selected and passaged on fresh YMG agar media. After repeated passaging, strains were tested on YMG-Hyg and YMG-5FC agar media. Four of the 130 strains were 5-FC-resistant with a Hyg-sensitive phenotype (Table 3). No hph amplicon was observed in one of the four Hyg-sensitive strains (Fig. 2c). Therefore, transient hph expression can be used to select the transformants under the Cas9 expression.

Marker-free genome editing by transient expression of Cas9, gRNA, and hph. a Schematic diagram of marker-free genome editing. b The plasmid map of pCcPef3-126-fcy1sg2. pUC ori: origin for Escherichia coli, KmR: kanamycin resistance gene, AbHSP26_ter: terminator from hsp26 gene in Agaricus bisporus. NLS: Nuclear localization signal, fcoCas9: Fungal and plant codon optimized Cas9 [10], CcEF3_pro: Promoter from ef3 gene in Coprinopsis cinerea, CcU6_pro: U6 promoter from C. cinerea, CctubB_pro or ter: Promoter or terminator from tubulin gene in C. cinerea, hph: hygromycin phosphotransferase. c PCR amplification of hph fragment using primer set hph_F and hph_R. Lane M, 1-kb ladder (Nippon gene); lane P, pPHT1 (positive control); lane W, parental strain PC9 (negative control); lanes 1–6, Pef3-1–6 strains. HygR: hygromycin B resistant, HygS: hygromycin B sensitive
Insertion of cas9 and genome editing of this Hyg-sensitive strain (Pef3-5) was confirmed. First, the cas9 amplicon was not observed following the colony PCR of the Pef3-5 strain (Fig. 3a). Next, amplification of a targeted site of gRNA in the fcy1 gene displayed a longer amplified fragment in Pef3-5 than in the wild-type strain (Fig. 3b, c). Sanger sequencing revealed an inserted sequence at the target site of the gRNA in the fcy1 gene (Fig. 3d, e). The first 37-bp could not be identified for sequence origin. Most of this fragment perfectly matched a small portion of the hph expression cassette. These results suggest that genome editing of the fcy1 gene in the Pef3-5 strain is mediated by the transient expression of Cas9, gRNA, and hph.

Confirmation of transient Cas9 expression and genome editing. a PCR amplification of cas9 gene fragment using primer sets Cas9check_F2 and Cas9check_R2. Lane M, 1-kb ladder (Nippon gene); lane P, pCcPef3-126-fcy1sg2 (positive control); lane W, parental strain PC9 (negative control); lanes1, Pef3-5. b A schematic diagram of the fcy1 loci in the PC9 parental strain with gRNA recognition sites. The dash lines indicate the region amplified by genomic PCR. Black arrows display the primer pairs used for the PCR experiments and Sanger sequencing. c PCR amplification of a target site of gRNA in the fcy1 using primer set TB9 and TB10. Lane M, 1-kb ladder (Nippon gene); lane W, parental strain PC9; lanes1, Pef3-5. d DNA sequencing to identify mutations of fcy1 in Pef3-5 strain. For highlights in the nucleotide sequence: yellow shades indicate gRNA, green shades indicate a protospacer adjacent motif (PAM) sequence, and dash lines indicate deletion by insertion of a plasmid sequence. e Insertion sequence in fcy1 locus of Pef3-5 strain revealed by Sanger sequencing. For highlights in the nucleotide sequence: blue shades indicate a sequence derived from hph, magenta shades indicate a sequence derived from CcTub_ter, and under lines indicate sequence derived from pCcPef3-126-fcy1sg2
This is the first example of marker-free genome editing by transient Cas9, gRNA, and hph expression in basidiomycetes. As cas9 is not integrated into the genome of the resulting strain (Fig. 3), our system should reduce off-target effects by avoiding constitutive expression of the CRISPR/Cas9 machinery. This editing method is expected to be suitable for creating genetically stable mutants.
This system requires a selectable marker only for initial selection of transformants. The marker is eventually lost in some transformants after repeated transfers. Hence, it is possible to repeatedly use selection markers for further genome editing using the method described in this study. This is the most important potentially novel advantage relative to other CRISPR/Cas9 protocols, including the ribonucleoprotein (RNP)-based genome-editing system. A pre-assembled RNP complex of Cas9 protein with in vitro transcribed gRNAs is an established method for performing gene mutation/disruption in eukaryotes without the possibility of transgene integration [26, 27]. In this case, the entire set required for genome editing is introduced directly into the host cell. After its introduction, these components are expected to cause genome editing and are eventually degraded by intracellular proteases and nucleases. An RNP-dependent CRISPR/Cas9 was successfully introduced into P. ostreatus in our previous research [12]. Although it is expected to be a genome-editing tool without any transgene introduction, there is no way to select RNP transformants unless the target gene functions as a selectable marker for genome editing. Therefore, donor DNA with a selection marker to be integrated into the host genome is usually used for direct introduction of Cas9 RNP [26]. Compared to this method, our transient expression system allows initial selection and genome editing of transformants via temporal expression of Cas9, gRNA, and the selection marker, and no transgene integration can be expected by further screening.
Generally, the frequency of homologous recombination in agaricomycetes is very low [16, 28], which may be due to high NHEJ activity. To effectively carry out gene targeting using conventional methods in agaricomycetes, isolation of a strain lacking the NHEJ pathway is essential. However, it is not easy to isolate such strains when ectopic incorporation of the introduced DNA occurs randomly. With our new protocol for transient CRISPR/Cas9-directed genome editing, multiple gene disruptions can be achieved in wild-type strains without isolating NHEJ-deficient strains. Therefore, this method is useful for editing multiple genes in many fungal species. Furthermore, using knock-in constructs, the variation of genome editing may be expanded largely using this method. To improve efficiency and reduce unexpected transgene integration in the chromosome, further technical improvements are being investigated.
Conclusion
In this study, transient expression of hph was used to select the transformants. Then, marker-free genome editing was achieved by the transient expression of Cas9, gRNA, and hph. Our new genome-editing system is expected to acquire fewer off-target effects by CRISPR/Cas9 and allow the repeated use of the selection marker to investigate the functions of multi-copy genes such as ligninolytic enzyme genes in the future.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- CRISPR:
-
Clustered regularly interspaced short palindromic repeat
- Cas9:
-
CRISPR-associated protein 9
- gRNA:
-
Guide RNA
- Hyg:
-
Hygromycin B
- hph :
-
Hygromycin phosphotransferase
- VP:
-
Versatile peroxidase
- MnP:
-
Manganese peroxidase
- fcy1 :
-
Cytosine deaminase
- 5-FC:
-
5-Fluorocytosine
- RNP:
-
Ribonucleoprotein
- NHEJ:
-
Non-homologous end joining
- YMG:
-
Yeast and malt extract with glucose
- PCR:
-
Polymerase chain reaction
- PAM:
-
Protospacer adjacent motif
References
Tyurin AA, Suhorukova AV, Kabardaeva KV, Goldenkova-Pavlova IV (2020) Transient gene expression is an effective experimental tool for the research into the fine mechanisms of plant gene function: advantages, limitations, and solutions. Plants 9:1187. https://doi.org/10.3390/plants9091187
Fus-Kujawa A, Prus P, Bajdak-Rusinek K, Teper P, Gawron K, Kowalczuk A, Sieron AL (2021) An overview of methods and tools for transfection of eukaryotic cells in vitro. Front Bioeng Biotechnol 9:701031. https://doi.org/10.3389/fbioe.2021.701031
Munoz-Rivas A, Specht CA, Drummond BJ, Froeliger E, Novotny CP, Ullrich RC (1986) Transformation of the basidiomycete Schizophyllum commune. Mol Gen Genet 205(1):103–106. https://doi.org/10.1007/BF02428038
Binninger DM, Skrzynia C, Pukkila PJ, Casselton LA (1987) DNA-mediated transformation of the basidiomycete Coprinus cinereus. EMBO J 6(4):835–840. https://doi.org/10.1002/j.1460-2075.1987.tb04828.x
van de Rhee MD, Graça PMA, Huizing HJ, Mooibroek H (1996) Transformation of the cultivated mushroom, Agaricus bisporus, to hygromycin B resistance. Mol Gen Genet 250(3):252–258. https://doi.org/10.1007/BF02174382
Sato T, Yaegashi K, Ishii S, Hirano T, Kajiwara S, Shishido K, Enei H (1998) Transformation of the edible basidiomycete Lentinus edodes by restriction enzyme-mediated integration of plasmid DNA. Biosci Biotechnol Biochem 62(12):2346–2350. https://doi.org/10.1271/bbb.62.2346
Honda Y, Matsuyama T, Irie T, Watanabe T, Kuwahara M (2000) Carboxin resistance transformation of the homobasidiomycete fungus Pleurotus ostreatus. Curr Genet 37(3):209–212. https://doi.org/10.1007/s002940050521
Honda Y, Tanigawa E, Tsukihara T, Nguyen DX, Kawabe H, Sakatoku N, Watari J, Sato H, Yano S, Tachiki T, Irie T, Watanabe T, Watanabe T (2019) Stable and transient transformation, and a promoter assay in the selective lignin-degrading fungus Ceriporiopsis subvermispora. AMB Expr 9(1):92. https://doi.org/10.1186/s13568-019-0818-1
Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9(1):1911. https://doi.org/10.1038/s41467-018-04252-2
Sugano SS, Suzuki H, Shimokita E, Chiba H, Noji S, Osakabe Y, Osakabe K (2017) Genome editing in the mushroom-forming basidiomycete Coprinopsis cinerea, optimized by a high-throughput transformation system. Sci Rep 7(1):1260. https://doi.org/10.1038/s41598-017-00883-5
Boontawon T, Nakazawa T, Inoue C, Osakabe K, Kawauchi M, Sakamoto M, Honda Y (2021) Efficient genome editing with CRISPR/Cas9 in Pleurotus ostreatus. AMB Expr 11(1):30. https://doi.org/10.1186/s13568-021-01193-w
Boontawon T, Nakazawa T, Xu H, Kawauchi M, Sakamoto M, Honda Y (2021) Gene targeting using pre-assembled Cas9 ribonucleoprotein and split-marker recombination in Pleurotus ostreatus. FEMS Microbiol Lett 368(13):fnab080. https://doi.org/10.1093/femsle/fnab080
Weng C, Peng X, Han Y (2021) Depolymerization and conversion of lignin to value-added bioproducts by microbial and enzymatic catalysis. Biotechnol Biofuels 14(1):84. https://doi.org/10.1186/s13068-021-01934-w
Nagy LG, Riley R, Bergmann PJ, Krizsán K, Martin FM, Grigoriev IV, Cullen D, Hibbett DS (2017) Genetic bases of fungal white rot wood decay predicted by phylogenomic analysis of correlated gene-phenotype evolution. Mol Biol Evol 34(1):35–44. https://doi.org/10.1093/molbev/msw238
Knop D, Yarden O, Hadar Y (2015) The ligninolytic peroxidases in the genus Pleurotus: divergence in activities, expression, and potential applications. Appl Microbiol Biotechnol 99(3):1025–1038. https://doi.org/10.1007/s00253-014-6256-8
Salame Tomer M, Knop D, Tal D, Levinson D, Yarden O, Hadar Y (2012) Predominance of a versatile-peroxidase-encoding gene, mnp4, as demonstrated by gene replacement via a gene targeting system for Pleurotus ostreatus. Appl Environ Microbiol 78(15):5341–5352. https://doi.org/10.1128/AEM.01234-12
Salame Tomer M, Knop D, Levinson D, Yarden O, Hadar Y (2013) Redundancy among manganese peroxidases in Pleurotus ostreatus. Appl Environ Microbiol 79(7):2405–2415. https://doi.org/10.1128/AEM.03849-12
Salame TM, Knop D, Levinson D, Mabjeesh SJ, Yarden O, Hadar Y (2014) Inactivation of a Pleurotus ostreatus versatile peroxidase-encoding gene (mnp2) results in reduced lignin degradation. Environ Microbiol 16(1):265–277. https://doi.org/10.1111/1462-2920.12279
Matsunaga Y, Ando M, Izumitsu K, Suzuki K, Honda Y, Irie T (2017) A development and an improvement of selectable markers in Pleurotus ostreatus transformation. J Microbiol Methods 134:27–29. https://doi.org/10.1016/j.mimet.2017.01.007
Larraya Luis M, Pérez G, Peñas María M, Baars Johan JP, Mikosch Thomas SP, Pisabarro Antonio G, Ramírez L (1999) Molecular karyotype of the white rot fungus Pleurotus ostreatus. Appl Environ Microbiol 65(8):3413–3417. https://doi.org/10.1128/AEM.65.8.3413-3417.1999
Nakazawa T, Tsuzuki M, Irie T, Sakamoto M, Honda Y (2016) Marker recycling via 5-fluoroorotic acid and 5-fluorocytosine counter-selection in the white-rot agaricomycete Pleurotus ostreatus. Fungal Biol 120(9):1146–1155. https://doi.org/10.1016/j.funbio.2016.06.011
Rao PS, Niederpruem DJ (1969) Carbohydrate metabolism during morphogenesis of Coprinus lagopus (sensu Buller). J Bacteriol 100(3):1222–1228. https://doi.org/10.1128/jb.100.3.1222-1228.1969
Cummings WJ, Celerin M, Crodian J, Brunick LK, Zolan ME (1999) Insertional mutagenesis in Coprinus cinereus: use of a dominant selectable marker to generate tagged, sporulation-defective mutants. Curr Genet 36(6):371–382. https://doi.org/10.1007/s002940050512
Nguyen DX, Nakazawa T, Myo G, Inoue C, Kawauchi M, Sakamoto M, Honda Y (2020) A promoter assay system using gene targeting in agaricomycetes Pleurotus ostreatus and Coprinopsis cinerea. J Microbiol Methods 179:106053. https://doi.org/10.1016/j.mimet.2020.106053
Izumitsu K, Hatoh K, Sumita T, Kitade Y, Morita A, Tanaka C, Gafur A, Ohta A, Kawai M, Yamanaka T, Neda H, Ota Y (2012) Rapid and simple preparation of mushroom DNA directly from colonies and fruiting bodies for PCR. Mycoscience 53(5):396–401. https://doi.org/10.1007/S10267-012-0182-3
Wang Q, Coleman JJ (2019) Progress and challenges: development and implementation of CRISPR/Cas9 technology in filamentous fungi. Comput Struct Biotechnol J 17:761–769. https://doi.org/10.1016/j.csbj.2019.06.007
Gong Z, Cheng M, Botella JR (2021) Non-GM genome editing approaches in crops. Front Genome Ed 3:817279. https://doi.org/10.3389/fgeed.2021.817279
Nakazawa T, Ando Y, Kitaaki K, Nakahori K, Kamada T (2011) Efficient gene targeting in ΔCc.ku70 or ΔCc.lig4 mutants of the agaricomycete Coprinopsis cinerea. Fungal Genet Biol 48(10):939–946. https://doi.org/10.1016/j.fgb.2011.06.003
Acknowledgements
We thank Professor Antonio Gerardo Pisabarro (Public University of Navarra) for providing the P. ostreatus strain PC9.
Funding
This work was supported in part by the Grants-in-Aid for Scientific Research [KAKENHI 18H02254, 18KK0178, 19K22332, and 21K18224 to YH], JSPS Bilateral Program [JPJSBP 120208402 and 120209920 to YH], and the Kyoto University Foundation [to MK].
Author information
Authors and Affiliations
Contributions
MK, TI and YH conceived and designed the study. HU, DK, MK, and MH performed the experiments. HU, DK and MK drafted the manuscript. HU, DK, and MK performed analyses. TN, MS, KI, TS, TI, and YH provided the editorial suggestions and revisions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Koshi, D., Ueshima, H., Kawauchi, M. et al. Marker-free genome editing in the edible mushroom, Pleurotus ostreatus, using transient expression of genes required for CRISPR/Cas9 and for selection. J Wood Sci 68, 27 (2022). https://doi.org/10.1186/s10086-022-02033-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s10086-022-02033-6