
Abstract
Background
Helicobacter pylori is an important agent of gastroduodenal disease in Africa and throughout the world. We sought to determine an optimum method for genotyping H. pylori strains from children and adults in The Gambia, West Africa.
Results
Virulence genes were amplified in 127 of 190 cases tested (121 adults and 6 children); each of 60 bacterial cultures, and 116 from DNA extracted directly from biopsies. The proportion of biopsies that were cagA+, the ratio of vacAs1/s2, and vacAm1/m2, and the proportion of mixed strain populations in individual subjects changed with age. Strains lacking virulence cagA and vacA genes and with apparently homogeneous (one predominant strain) infections were more common among infants than adults.
Conclusions
In order to detect the range of bacterial genotypes harbored by individual patients, direct PCR proved slightly superior to isolation of H. pylori by biopsy culture, but the techniques were complementary, and the combination of both culture and direct PCR produced the most complete picture. The seemingly higher virulence of strains from adult than infant infections in The Gambia merits further analysis.
Similar content being viewed by others


Background
Helicobacter pylori chronically infects over 50% of people worldwide, causes gastritis and sometimes gastric or duodenal ulceration, and increases the risk of gastric cancer [1, 2]. Infection also contributes to other maladies such as malnutrition among the very poor, iron deficiency anemia, and susceptibility to other food and water borne pathogens, especially in developing countries, including The Gambia [3, 4]. The prevalence of H. pylori infection is particularly high in developing countries including The Gambia [5–7]. H. pylori, because it is a fastidious micro-aerobic bacterium, it is technically difficult to grow and maintain for molecular biologic research in poorly resourced laboratories in Africa. These challenges coupled with the uniqueness of genotypes of African strains and special features of human physiology and environment in this continent limit our understanding of the spectrum of H. pylori-associated diseases and how this is affected by bacterial genotype in Africa [8, 9]. So extensive efforts have been made to determine an optimum method for PCR-based genotyping of H. pylori[10–13]. To more effectively investigate the influence of H. pylori genotype on associated diseases in a West African setting, this study sought to determine an optimum method for PCR-based genotyping of H. pylori in The Gambia, West Africa.
Results
A total of 169 biopsy samples from adult subjects, and 21 from infants were investigated for H. pylori infections by both culture and PCR of DNA obtained directly from biopsies. 89/169 (52.6%) adults seemed to be culture positive. Pure H. pylori cultures were obtained from only 63 of them, but not the other 26, primarily because of overgrowth by contaminants despite inclusion of multiple antibiotics in the culture medium or bacterial cells failing to survive further subculture. Direct PCR from adult biopsies indicated that 164/169 (97%) were positive for Hp16s (table 1). The DNA extracts from the remaining 5 biopsies were H. pylori negative with Hp16s and did not amplify for any of the genes tested, even though they were culture positive. These five samples were tested for PCR inhibitors by spiking them with DNA from a known positive. The spiked samples were also all negative after PCR (data not shown), which implies the presence of a potent inhibitor, possibly a ribonuclease. Virulence gene data were obtained by direct PCR from both biopsies and cultures in 60 cases and these data are used for the comparisons in table 2.
Amongst the 80 culture negative adult subjects for whom Hp16s was positive, amplification of some or all virulence genes was only achieved in 20 cases. It is not yet known if these strains lacked virulence genes, were divergent in primer binding sequences, or the bacterial density was so low that amplification was possible only with the most general of primer combinations, such as Hp16s. The remaining 60 samples did not show amplification for any of the genes tested despite a positive response to Hp16s.
Amongst 21 infants, 8 were Hp16s positive and pure H. pylori cultures were obtained successfully from six of them. We succeeded in amplifying virulence gene sequences only from the 6 culture positive children. Direct PCR of the biopsies from the other 13 children were either negative (n = 10) or not done (n = 3).
Virulence gene amplification was successful in 127 (121 adults and 6 children) cases. A comparison of the products that were indicative of cagA (Figure 1), cag emptysite (Figure 2), vacAs alleles (Figure 3), vacAm alleles (Figure 4), iceA1 (Figure 5) and iceA2 (Figure 6) between both methods for detecting H. pylori is summarized for 60 samples for which sufficient amplified DNA was obtained for this further analysis. The proportion of samples that were cagA+ve with DNA from biopsies and from culture was similar, 58.3% and 61.7% respectively. The success in amplification of vacAs1/s2 (s1 = toxigenic vs s2 = non-toxigenic), vacAm1/m2 and iceA1 alleles was similar from cultures and corresponding biopsies, and agreements between genotypes inferred using DNAs directly from these two sources was good for both cagA and m1, m2 alleles of vacA, moderate for s1, s2 alleles of vacA, and poor for iceA. The poor agreement in the iceA analysis stemmed from the many classified only as iceA2 by PCR from bacterial culture but iceA1 and iceA2 by biopsy which could have been due to the fact that certain bacterial strains in a mixed infection grew much better than others in culture. In direct PCR up to 16.7% of culture positive biopsies failed to amplify DNA for individual alleles.

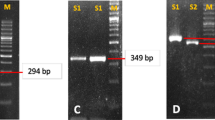
PCR inferred results of cagA gene. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of cagA gene. Lane M is a 100 bp ladder (Biolabs, UK); lanes 1, 2, 3 and 5 showed PCR products (349 bp) of cagA genes, lane 4 is cagA negative.

PCR inferred results of cag emptysite. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of cag emptysite. Lane M is a 100 bp ladder (Biolabs, UK), lanes 1, 2 and 4 showed PCR products of 535 bp indicating the presence of cag emptysite, lanes 3 and 5 were cag emptysite negative.

PCR inferred results of signal region of vacA gene. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of s1 and s2 allelles of vacA gene. Lane M is a 100 bp ladder (Biolabs, UK), lane 1 and 2 showed the presence of s1 (259 bp), lane 3 is both s1 and s2 positive and lane 4 and 5 are s2 (289 bp) positive.

PCR inferred results of mid region of vacA gene. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of m1 and m2 allelles of vacA gene. Lane M is a 100 bp ladder (Biolabs, UK), lanes 1, 2 and 3 are m1 positive; lanes 4 and 5 showed the presence of m2.

PCR inferred results iceA1. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of iceA1 gene. Lane M is a 100 bp ladder (Biolabs, UK), lanes 1, 2 and 5 showed the presence of 297 bp of iceA1, lanes 3 and 4 are iceA1 negative.

PCR inferred results iceA2. 1.5% gel electrophoresis of H. pylori genotypes showing PCR results of iceA2. Lane M is a 100 bp ladder (Biolabs, UK), lanes 1 and 2 showed the presence of 334 bp of iceA2 and lanes 3 and 4 were iceA2 positive of 229 bp, lane 5 was iceA2 negative.
The proportion of biopsies that were cagA+, the proportion of vacAs1, and vacAm1, and the proportion of mixed cultures from individual subjects varied with age. Table 3 is a summary of all PCR results, including samples obtained from cultures and from direct PCR on biopsies (127 in total). If subjects were positive by both techniques, only the biopsy amplified sample was included in this analysis. None of the young children had mixed cultures with relation to cagA, vacAs or vacAm alleles. Young children also exhibited lower levels of the toxigenic genes than any of the adult groups. This difference was only statistically significant (P≤0.02) when isolates obtained from children were compared with those from adults aged less than 60 years for cagA and s1 allele of vacA, and when compared with isolates from adults aged 41-59 years for m1 region of vacA. However, the sample size in children was small and therefore the difference between children and adults should be interpreted with caution. The prevalence of virulence genes was age-dependent. For cagA, vacAs and vacAm the virulent genotype was most common among the 30-40 year age group and less common in younger and older age groups. This association was statistically significant (p < 0.05) for cagA and vacAs and not for the mid region of vacA gene and iceA alleles (p>0.05, table 3). Only 1 elderly subject (70 years) was found to have mixed colonization with vacAs1/s2. The situation with iceA was more complicated, with a large number of individuals exhibiting mixed iceA1/iceA2 colonization.
Discussion
In this study, we describe the comparison between results obtained from direct PCR to detect H. pylori from gastric biopsies in West Africa, compared to PCR of bacterial isolates obtained from the same set of gastric biopsies. Both techniques produced different success rates, as set out in table 1 and both failed to detect H. pylori in a significant proportion of infections. We agree with Park et al [14] in that direct PCR can produce inconsistent results, and tend to underestimate the prevalence of specific virulence factors (table 2). However, in this study, we detected a good consistency of genotypes between both techniques consistent with what was reported in a similar study [10].
Our data differs in that we experienced considerably greater difficulty in obtaining pure subcultures of H. pylori from gastric biopsies than Park, with a consequently higher failure rate. We have been involved in studies cultivating H. pylori from gastric biopsies from populations throughout the world, and it is our personal observation that sub-culture failure is a particular problem amongst West African isolates, as encountered in the present study. The reasons for this are not immediately apparent.
As a consequence of this problem, not all biopsies from which virulence factor DNA was amplified yielded a primary isolation of H. pylori, and there was a significant loss of isolates at subculture. PCR from subcultures gave higher rates of mixed colonization for cagA and vacA genes than direct PCR of biopsies, in contrast to the situation reported elsewhere with higher culture success rates [14]. This may have been due to artifact, either by enhancement of a minor strain from within the stomach, or due to modification of genome during culture [15]. In our hands, therefore, direct PCR produced more positive results, gave rise to fewer concerns about the development of artifact, and was more rapid and convenient.
Our data also indicate that there may be PCR inhibitors or potent nucleases in some gastric biopsies. This is consistent with findings in similar studies [12, 14]. Their occasional presence and the underestimated prevalence of specific virulence factors by direct PCR illustrate that culture can be a useful complement to direct PCR for studies in which complete ascertainment of H. pylori virulence factor genotypes, including mixed colonization, is desired.
We observed a difference in predominant genotype with subjects' age. Young children produced isolates that were more likely to be cagA-ve, and VacAs2m2, in contrast to adults who were more likely to harbor cagA+ve VacAs1m1 isolates. Children were also less likely to have mixed populations of H. pylori strains, which may relate to children aged 18 to 31 months being relatively recently colonized by H. pylori, compared to older individuals. The strains of H. pylori discovered in adult stomachs, at ages when typical H. pylori associated diseases develop, may be genotypically distinct from the original strains that first colonized young Gambian children. This could be due to recombination of the H. pylori genome over the course of decades [16, 17] and/or re-exposure to novel strains, with more pathogenic strains circulating predominantly amongst adults.
Conclusion
In order to detect the range of bacterial genotypes harbored by individual patients, direct PCR proved slightly superior to isolation of H. pylori by biopsy culture in our hands, but the techniques were complementary to each other, and the use of both together produced the most complete picture. Despite the lower success rate and greater cost of H. pylori culture relative to PCR directly from biopsies, culturing H. pylori is still important for antibiotic susceptibility tests that could guide therapy and other phenotypic tests such as bacterial adherence, cagA and vacA action on mammalian cells, expression of other colonization and virulence traits for which PCR alone is unsuitable.
Methods
Patients
Ethical approval of study protocols was obtained from the joint Gambian Government MRC Ethical Committee and from The London School of Tropical Medicine and Hygiene.
169 gastric antral biopsies were obtained from adult subjects (50 female and 71 male) undergoing routine diagnostic endoscopy after obtaining informed consent. These subjects were consecutive patients attending the MRC endoscopy clinic for whom the endoscopist decided it was appropriate to take biopsies for research as well as clinical purposes. Patients with severe oesophago-gastroduodenal disease, including gastro-oesophageal varices or gastric cancer, were therefore not included in the study.
In addition, gastric biopsies were obtained, after informed parental consent, from 21 children aged 18 to 31 months, who were undergoing endoscopic small bowel biopsy because of suspected enteropathy.
The biopsies were immediately stored in Brain Heart Infusion (BHI) broth containing 20% glycerol and transported in ice to the laboratory for processing or stored at -70°C until used.
Culture
Endoscopic biopsies were spread on the surface of selective Columbia-blood agar (Unipath, Basingstoke, UK) supplemented with 10% sheep blood (TCS Biosciences, UK), 2% vitox (Unipath, Basingstoke, UK) and the following antibiotics: trimethoprim (5 μg/ml), vancomycin (6 μg/ml), polymixin B (10 μg/ml), bacitracin (200 μg/ml), nalidixic acid (10 μg/ml), and an antifungal amphotericin B (8 μg/ml) [18]. The inoculated plates were incubated in a micro-aerobic atmosphere at 37°C for 5-7 days. Isolation and identification of H. pylori was made by colony morphology, Gram stain, oxidase, urease and catalase activity. Strains were preserved in BHI broth containing 20% glycerol and stored at -70°C.
DNA extraction from cultures
DNA was prepared by harvesting a confluent growth of pooled H. pylori population from agar media and extracted using a commercial kit (QiagenR DNA Mini Kit, UK) as per manufacturer's guidelines. The DNA was stored at -20°C until used for gene amplification.
DNA extraction directly from biopsies
Total genomic DNA was extracted from the biopsy samples by using a combination of the QIAamp DNA isolation kit (Qiagen, UK) and a bead-beater method. Briefly, biopsies were lysed in 180 μl of QIAamp ATL buffer and 20 μl of proteinase K for 1 h at 56°C. Glass beads of different diameters (0.1 mm, 0.5 mm and 1 mm, Sigma) were added, and samples were homogenized in a FastPrep FP120 bead-beater (Bio101, Savant Instruments) for 30 sec at 4 m/s and incubated for an additional hour at 56°C. 200 μl of AL buffer were added to the lysate and samples were incubated for 30 min at 70°C. After the addition of 200 μl absolute ethanol, lysates were purified over a QIAamp column as specified by the manufacturer.
PCR amplification of H. pylori 16s rRNA
PCR was performed on extracted DNA from biopsies and also from cultures using H. pylori 16s rRNA specific PCR ["Hp16s"] as previously described [19] under the following conditions: 35 cycles of 95°C for 30s, 60°C for 30s and 72°C for 30s and an extension time of 72°C for 5 min. The amplified genes were detected by electrophoresis in a 1.5% agarose gel with ethidium bromide (500 ng/ml) and bands visualized using Gel Doc 2000 (Bio-Rad laboratories, Milan, Italy).
PCR to detect genotypes
PCR was performed to detect cagA, vacA genes, iceA1 and iceA2 using previously described methods [20], under the following general conditions: 30 cycles of 94°C for 1 min, 55°C for 1 min and 72°C for 1 min. The amplified genes were detected by electrophoresis in a 1.5% agarose gel with ethidium bromide and bands were visualized using Gel Doc 2000 (Bio-Rad laboratories, Milan, Italy). The primers used are listed in table 4.
Statistics
Percentage agreement was calculated to compare H. pylori genotypes obtained by PCR performed directly on gastric biopsies with the genotypes obtained by PCR of DNA extracted from bacteria cultured. In addition, we report the kappa statistic which allows for chance agreement (kappa = 0 corresponds to no agreement beyond that expected by chance and kappa = 1 represents perfect agreement).
We studied the prevalence of genotypes within different age categories. The null hypothesis of no association between prevalence and age was tested using Fisher's exact test.
References
Mbulaiteye SM, H M, El-Omar EM: Helicobacter pylori associated global gastric cancer burden. Front Biosci. 2009, 14: 1490-504.
Wu IC, W D, Yu FJ, Wang JY, Kuo CH, Yang SF, Wang CL, Wu MT: Association between Helicobacter pylori seropositivity and digestive tract cancers. World J Gastroenterol. 2009, 15 (43): 5465-71. 10.3748/wjg.15.5465.
Akcam M: Helicobacter pylori and micronutrients. Indian Pediatr. 2010, 47 (2): 119-26. 10.1007/s13312-010-0017-2.
Fernández-Bañares F, M H, Forné M: A short review of malabsorption and anemia. Indian Pediatr. 2010, 47 (2): 119-26. 10.1007/s13312-010-0017-2.
Goodman KJ, C P, Tenganá Aux HJ, Ramírez H, DeLany JP, Guerrero Pepinosa O, López Quiñones M, Collazos Parra T: Helicobacter pylori infection in the Colombian Andes. a population-based study of transmission pathways. Am J Epidemiol. 1996, 144: 290-9.
Thomas JE, D A, Harding M, Coward WA, Cole TJ, Weaver LT: Helicobacter pylori colonization in early life. Pediatr Res. 1999, 45: 218-223. 10.1203/00006450-199902000-00010.
Weaver L: Aspects of Helicobacter pylori Infection in the developing and developed world. Helicobacter pylori infection, nutrition and growth of West African infants. Transactions Royal Soc Trop Med Hygiene. 1995, 89: 347-50. 10.1016/0035-9203(95)90002-0.
Campbell DI, W B, Thomas JE, Figura N, Telford JL, Sullivan PB: The African enigma: low prevalence of gastric atrophy, high prevalence of chronic inflammation in West African adults and children. Helicobacter. 2001, 6: 263-267. 10.1046/j.1083-4389.2001.00047.x.
Holcombe C: Helicobacter pylori - the African enigma. Gut. 1992, 33: 429-431. 10.1136/gut.33.4.429.
Santanu Chattopadhyay, P R, Ramamurthy T, Abhijit Chowdhury, Amal Santra, Dhali GK, Bhattacharya SK, Berg Douglas E, Balakrish Nair, Mukhopadhyay Asish K: Multiplex PCR Assay for Rapid Detection and Genotyping of Helicobacter pylori Directly from Biopsy Specimens. J Clin Microbiol. 2004, 42 (6): 2821-2824. 10.1128/JCM.42.6.2821-2824.2004.
Lim Chang-Young, Cho Myung-Je, Chang Myung-Woong, Kim Seok-Yong, Myong Na-Hye, Lee Woo-Kon, Rhee Kwang-Ho, Kook Yoon-Hoh: Detection of Helicobacter pylori in Gastric Mucosa of Patients with Gastroduodenal Diseases by PCR-Restriction Analysis Using the RNA Polymerase Gene (rpoB). J Clin Microbiol. 2003, 41 (7): 3387-3391. 10.1128/JCM.41.7.3387-3391.2003.
Thoreson A-CE, B M, Andersen LP, JÖrgensen F, Kiilerich S, Scheibel J, Rath J, Krogfelt KA: Helicobacter pylori detection in human biopsies: a competitive PCR assay with internal control reveals false results. FEMS Immunology and Medical Microbiology. 1999, 24: 201-208. 10.1111/j.1574-695X.1999.tb01283.x.
Smith SI, O K, Arigbabu AO, Cantet F, Megraud F, Ojo OO, Uwaifo AO, Otegbayo JA, Ola SO, Coker AO: Comparison of three PCR methods for detection of Helicobacter pylori DNA and detection of cagA gene in gastric biopsy specimens. World J Gastroenterol. 2004, 10 (3): 1958-1960.
Park Chang-Young, M K, Gutierrez Oscar, Graham David, Yamaoka Yoshio: Comparison of Genotyping Helicobacter pylori Directly from Biopsy Specimens and Genotyping from Bacterial Cultures. J Clin Microbiol. 2003, 41: 3336-3338. 10.1128/JCM.41.7.3336-3338.2003.
Hua J, HC N, Yeoh Guan Khay, Ho Bow: Predominance of a Single Strain of Helicobacter pylori in Gastric Antrum. Helicobacter. 1999, 4: 28-32. 10.1046/j.1523-5378.1999.09043.x.
Danon SJ, BJ L, Mankoski Raymond, Eaton Kathryn: RFLP and RAPD Analysis of In Vivo Genetic Interactions Between Strains of Helicobacter pylori. Helicobacter. 1998, 3: 254-259. 10.1046/j.1523-5378.1998.08010.x.
Taylor NS, F J, Akopyants NS, Berg DE, Thompson N, Shames B, Yan LL, Fontham E, Janney F, Hunter FM, Correa P: Long-term colonization with single and multiple strains of Helicobacter pylori assessed by DNA fingerprinting. J Clin Microbiol. 1995, 33: 918-23.
Akada JK, O K, Dailidiene D, Dailide G, Cheverud JM, Berg DE: Helicobacter pylori tissue tropism: mouse-colonizing strains can target different gastric niches. Microbiology. 2003, 149: 1901-9. 10.1099/mic.0.26129-0.
Sheng-Ang Ho, JA H, Lewis Fraser, Secker Alison, Cross Debra, Mapstone Nicholas, Dixon Michael, Wyatt Judy, Tompkins David, Taylor Graham, Quirkel Philip: Direct Polymerase Chain Reaction Test for Detection of Helicobacter pylori in Humans and Animals. J Clin Microbiol. 1991, 29: 2543-2549.
Mukhopadhyay AK, D K, Jin-Yong Jeong, Datta Simanti, Ito Yoshiyuki, Chowdhury Abhijit, Chowdhury Sujit, Santra Amal, Bhattacharya Sujit, Azuma Takeshi, Balakrish Nair G, Berg Douglas: Distinctiveness of genotypes of Helicobacter pylori in Calcutta. India. J Bacteriol. 2002, 182: 3219-3227.
Acknowledgements
This work was supported in part by grant RO3-AI061308 from the US National Institutes of Health and The Medical Research Council Laboratories, The Gambia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests and the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' contributions
JET, RAA and DEB conceived the study. OS performed all the experiments, analysis and wrote the paper with JET, DEB and RAA. TC, JET, MT, RW collected all biopsies from subjects referred for clinical diagnoses. VT participated in consenting of patients and preparing the patients for endoscopy. CB was involved in the statistical analysis. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Secka, O., Antonio, M., Tapgun, M. et al. PCR-based genotyping of Helicobacter pylori of Gambian children and adults directly from biopsy specimens and bacterial cultures. Gut Pathog 3, 5 (2011). https://doi.org/10.1186/1757-4749-3-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1757-4749-3-5