
Abstract
Background
Genome-wide studies on autism spectrum disorders (ASDs) have mostly focused on large-scale population samples, but examination of rare variations in isolated populations may provide additional insights into the disease pathogenesis.
Methods
As a first step in the genetic analysis of ASD in Croatia, we characterized genetic variation in a sample of 103 subjects with ASD and 203 control individuals, who were genotyped using the Illumina HumanHap550 BeadChip. We analyzed the genetic diversity of the Croatian population and its relationship to other populations, the degree of relatedness via Runs of Homozygosity (ROHs), and the distribution of large (>500 Kb) copy number variations.
Results
Combining the Croatian cohort with several previously published populations in the FastME analysis (an alternative to Neighbor Joining) revealed that Croatian subjects cluster, as expected, with Southern Europeans; in addition, individuals from the same geographic region within Europe cluster together. Whereas Croatian subjects could be separated from a sample of healthy control subjects of European origin from North America, Croatian ASD cases and controls are well mixed. A comparison of runs of homozygosity indicated that the number and the median length of regions of homozygosity are higher for ASD subjects than for controls (p = 6 × 10-3). Furthermore, analysis of copy number variants found a higher frequency of large chromosomal rearrangements (>2 Mb) in ASD cases (5/103) than in ethnically matched control subjects (1/197, p = 0.019).
Conclusions
Our findings illustrate the remarkable utility of high-density genotype data for subjects from a limited geographic area in dissecting genetic heterogeneity with respect to population and disease related variation.
Similar content being viewed by others


Background
Autism Spectrum Disorders (ASD, MIM209850) are a severe neuropsychiatric disorders, primarily characterized by abnormalities in social behavior, communication and language, with patterns of restricted and repetitive interests [1]. Commonly associated symptoms include aggressive and self-injurious behaviors, anxiety, heightened sensitivity to stimuli, and seizures [2, 3]. Severity of symptoms in ASD can vary widely among cases, from individuals with mental retardation and no language production to relatively high-functioning individuals with normal to superior intelligence but significant difficulties with social interaction [4–6].
Multiple lines of evidence converge to suggest that ASDs represent the most heritable neurodevelopmental and psychiatric conditions. Evidence from twin and family studies suggests that the rate of autism in siblings of affected individuals is 2-6% [7], with 92% concordance of ASD in monozygotic twins and over 10% concordance in dizygotic twins [8]. Previous linkage and candidate gene studies have identified several chromosomal regions with autism susceptibility loci [3, 9–13]. A recent study identified a common genetic risk factor underlying ASD employing a genome-wide association strategy in 780 families (3,101 subjects) with affected children, and a second cohort of 1,204 affected subjects and 6,491 control subjects; these findings were replicated in an independent study cohort of 483 ASD families [14]. Furthermore, substantial progress towards the identification of genetic risk variants has come from recent characterization of structural variation (i.e., copy number variation or CNV). Studies of CNVs in a cohort of 859 ASD cases and 1,400 healthy control children revealed structural variants in previously reported ASD candidate genes (NRXN1 and CNTNAP4) and in multiple novel susceptibility genes encoding cell-adhesion molecules, including NLGN1 and ASTN2. Furthermore, CNVs observed in cases but not controls were found within and surroundings genes involved in the ubiquitin pathway (UBE3A, PARK2, RFWD2 and FBXO40) [15]. In a related study, we performed high-density genotyping and obtained data on 3,832 individuals from 912 multiplex ASD families in the Autism Genetic Research Exchange (AGRE) and 1,489 unrelated control subjects. Through prioritization of exonic deletions (eDels), we recovered genes in which structural variants were present in multiple unrelated probands but not unrelated controls. The 42 genes identified by this method include NRXN1, UBE3A, MADCAM1 and BZRAP1 [16]. Studies of large families with shared ancestry have reported several other autism loci including large, inherited, homozygous deletions in neuronal cell-adhesion genes, for example PCDH10 (protocadherin 10) [17]. These findings highlight the utility of "homozygosity mapping" and a need to search for additional large families across diverse populations.
Based on these breakthroughs and lessons learned from studies of other complex diseases (i.e. diabetes and breast cancer), it is becoming apparent that there is a need to assess large sample collections that number in the tens of thousands. Population structure poses a challenge for large-scale disease-association studies; mismatched ancestry between cases and controls in a genome-wide association studies is a potential source of spurious associations [17]. These needs and challenges bring together usually separated fields of genetics, epidemiology and studies of human diversity. Also, it is important to relate genetic findings found in patients from developed countries to the studies of diverse populations in less developed parts of the world. Currently DNA has been assembled from only a few thousand individuals with autism worldwide. Significant advances in genetic analyses of ASD in diverse populations will require the collection of samples across many populations using rapid and affordable screening and diagnostic tools.
Isolated populations have contributed to the discovery of genetic factors for many Mendelian disorders [18]. It has been argued that isolated populations may facilitate the identification of susceptibility loci for complex diseases because of a reduced genetic diversity, i.e. reduced genetic heterogeneity [19, 20]. Although several Croatian island populations have been described as genetic isolates, the Croatian population as a whole does not represent a population isolate as conventionally defined [21, 22]. However, Croatia is a small country with a low migration rate, in which large extended families often live in the same household or same neighbourhood and, therefore, is perfectly suited for family-based genetic studies. We expect that a subset of affected individuals may segregate a limited number of causative ASD susceptibility alleles (causal variants); in some cases these loci or disease-associated haplotypes identified in genetic isolates may differ from those in more genetically diverse populations, such as Europeans in North America. We also expect that a subset of rare variants associated with ASD susceptibility (SNPs and CNVs), which were identified in a large set of Europeans from North America, may be more prevalent in specific ethnic groups like the Croats. Therefore, to improve our understanding of global patterns of human genetic variation and set the stage for large-scale disease studies, efforts are underway to evaluate genetic variation using high-density genotype data for human populations worldwide [23–25].
With the intention to use the Croatian population in future genome-wide association studies, we performed a genome-wide high-density genotype pilot study of 103 ASD cases and 203 control samples to characterize the genetic structure of the population. Our analysis focuses on (1) the genetic diversity of the Croatian population, and its relationship to other populations, (2) the degree of relatedness of the population through the analysis of Runs of Homozygosity (ROHs), and (3) the distribution of large (>500 Kb) copy number variations, a major mechanism for structural variation in human genomes, and its possible association with ASD.
Results and Discussion
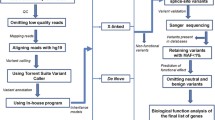
To understand the complexities of genomic architecture of ASDs, a highly heterogeneous group of disorders, and to assess the distribution and specificity of common and rare variants in different populations, we initiated genomic analysis of ASD and control subjects from Croatia. This cohort, consisting of 103 subjects (children and adults) with ASD and 203 adult control subjects, with male-to-female ratios of 3.7:1 (in ASD) and 2.6:1 (in controls), was originally recruited to study the role of hyperserotonemia in autism [26–28]. Patients with autism were evaluated by a child psychiatrist and diagnoses were based on DSM-IV-TR [29] criteria. Severity of behavioral symptoms was measured using the Childhood Autism Rating Scale (CARS) [30]. The degree of mental retardation (MR) was assessed according to the standardized intelligence or developmental tests, corresponding to the apparent developmental level of each individual. ASD in selected families occurs either in the absence of positive family history (sporadic) or in families with a high prevalence of other clinical psychiatric disorders distinct from ASD (such as anxiety, depression and phobias). During the clinical assessment and interviews, psychiatrists and geneticists did not notice any recognizable dysmorphic features in selected study subjects. DNA samples isolated from whole blood were genotyped with the Illumina HumanHap550 SNP array and 306 samples gave high quality data that were subjected to population structure and CNV analysis. See Table 1 for a summary of the genome-wide datasets used in this study.
To understand the genetic homogeneity of the Croatian cohort and its position in the worldwide human population, we used phylogenetic tree analysis, principal component analysis (PCA) (not shown), and multi-dimensional scaling (MDS), three tools commonly used in the study of human population genetic diversity [23, 24]. We used data from the Human Genome Diversity Project (HGDP), which consists of genome-wide SNP data of 1007 individuals from 51 populations, divided into 7 geographic regions. Of these individuals, 153 were from 8 populations from the European region. We plotted the results of MDS and tree analyses using control subjects from Croatia and the HGDP samples (Figure 1). All three methods show the same overall pattern emerging from previous HGDP studies: the relative positions of the major regions largely correlate with their relative geographical locations, and reflect the human migration history well. Humans migrated from Africa to the Middle East (Figure 1A-B); the first branching took place, and one group migrated to Europe, while the other migrated to South Asia. A second major branching then occurred, with one migration to America (through Alaska), the other countinuing to East Asia and eventually Oceania. As expected, the Croatia control samples are entirely contained within the European samples. A focused analysis of Europeans in the HGDP panel (Figure 1C-D) with the Croatia control samples situated in the middle of phylogeny or MDS plots shows the relationship of this subpopulation to other European populations. The Sardinians and the Basques seem to be the farthest, with Italian samples placed in between them and the Croatian controls; the other subpopulation that is separate from the Croatia controls is the Adygei. The Croatians are most similar to the Russians and the Orcadians. Both the PCA (not shown) and MDS plots show the relative diversity of the Croatian controls (indicated by the area spanned by the samples on the MDS and PCA plots) to be similar to the other populations. The boxplot of pairwise Identity by State (IBS) distances for European populations (Figure 2A) showed the Croatian control cohort has divergence similar to other European subpopulations (except Basques and Sardinians who are genetically more homogeneous), and smaller than the European population as a whole.

Population structure of Croatia Cohort. (A) FastME tree using IBS distance and (B) IBS distance multidimensional scaling (MDS) for Croatia control + HGDP, colored by HGDP region IDs. (C) FastME tree using IBS distance and (D) IBS distance multidimensional scaling (MDS) for Croatia Control + HGDP European Only, colored by HGDP subpopulation IDs.

Pairwise IBS analysis. (A) Box-and-whisker plot of pairwise IBS distances for Croatia Control, subpopulations of HGDP European Cohort, and the whole HGDP European Cohort. (B) Scatter plot of the first two coordinates of IBS distance multidimensional scaling (MDS) for Croatia control, case, and NINDS control cohorts.
We performed MDS on the Croatian controls and cases, as well as NINDS (National Institute of Neurological Disorders and Stroke) control cohorts to check the relationships between the three cohorts (Figure 2B), and found that the Croatian control and cases are well mixed but separate from the NINDS controls. Randomization tests by shuffling between Croatian control and NINDS controls showed the median pairwise IBS (identity-by-state) distance across the two subsets (0.28) is significantly higher than expected by chance (p = 0.00273, see Figure 3); this agrees with the MDS visualization that the two cohorts are genetically separate. Analysis of fixation index (FST) led to the same conclusion (see Table 2 for top regions with large FST). In summary, the Croatian and the NINDS cohorts have significant genetic difference, and care must be taken to correct for population stratification in combining the two datasets for association studies.

Analysis of median and mean cross-population pairwise IBS distances between Croatia Control and NINDS. (A) Observed median IBS distance and distribution of median IBS distances by random shuffling of population memberships; (B) observed mean IBS distance and distribution of median IBS distances by random shuffling of population memberships.
Mapping of large regions of homozygosity in pedigrees with shared ancestry has been employed in the discovery of autosomal recessive diseases associated with neurodevelopmental anomalies [31–33], and more recently, in the identification of novel genes associated with ASD, schizophrenia and Alzheimer's Disorder [17, 34, 35]. Although we recruited unrelated ASD cases and healthy controls, we analyzed genome-wide rates of homozygosity to estimate consanguinity and compared these rates to a heterogeneous control sample of Europeans from North America. It has been suggested that mapping of extended tracks of homozygosity may facilitate identification of low-frequency variants associated with complex diseases [36, 37]. We detected regions ("runs") of homozygosity (ROH) using the PLINK v1.05 software [38] and compared the extent of homozygosity in Croatia ASD cases, control subjects and NINDS controls. We computed the number of ROH segments, total ROH length, and average ROH length for the three subsets (Table 3). We detected subtle differences between the three groups for the median number of ROHs (Kruskal-Wallis test; p = 0.06), as well as average ROH length between Croatia controls and NINDS controls (Wilcoxon test; p = 0.053), with NINDS cohort showing shorter total ROH length than Croatia controls (Kruskal-Wallis test; p = 0.0056, higher due to the very large number of NINDS samples). However, the most significant pairwise difference is the total ROH length between Croatian cases Croatian controls (Wilcoxon test; p = 0.0065).
Motivated by the study demonstrating the positive correlation between runs of homozygosity and age in a North American cohort of European descent due to urbanization [35], we examined age correlation with ROH frequency and size in the 103 ASD subjects (Figure 4 and Table 4). We found only borderline significance between age and average ROH size, though the lack of significance may be due to the fact that the Croatia ASD cohort is 40 years younger (mean age is 21.5 y as opposed to 61.7 y in Nalls et al. [35]).

Scatter plots for ROH number and length versus Age of Subject. (A) Total ROH length (in Kb) versus Age; (B) Number of ROHs versus Age; (C) Average ROH size (in Kb) versus Age.
ROH regions were common or specific for a demographic group (Croatia vs. North America) or a disease status (ASD vs. controls). Although the longest ROH regions do not need to necessarily coincide with the disease susceptibility, we cannot exclude a possibility that recessive variants, such as single nucleotide or copy number variants within these regions contribute to disease susceptibility. To identify which regions contribute to the significant difference between Croatia case and control groups, we sorted the 7,740 endpoints of all autosomal ROHs across Croatia case and control groups, computed the significance of association between ROH status and case/control status by Fisher's test for each pair of adjacent ROH endpoints, and examined the significance of each such ROH interval where at least 5% (15) individuals have the ROH (Table 5). The most significant ROHs are in two chromosomal regions: chr2:82,040,863-82,677,336 (2p12) and chr6:29,481,192-29,719,410 (6p21.1) (Figure 5 and Figure 6). It is striking these two homozygosity regions are near several genes previously associated with neurological and psychiatric disorders. The chromosome 2 interval (636 Kb) is located downstream of CTNNA2 (alpha catenin) and LRRTM1 (leucine-rich repeat transmembrane neuronal protein 1), and contains a segment (between SNPs rs2685729 and rs2862972) significantly enriched in ASD subjects (top p = 2.5 × 10-3). The LRRTM1 gene is associated with left-handedness (patients with autism and many other psychiatric disorders have a higher tendency to be left-handed [39]) and may increase risk of schizophrenia [40, 41]. The chromosome 6 interval (238 Kb) is a gene-rich region located distal to the major histocompatibility complex. This ROH is shared by 17 ASD subjects (16.5%) and 19 controls (9.6%) spans 8 genes, including a cluster of olfactory receptors (OR11A1, OR10C1, OR2H1, OR2H2), the MOG locus, and the GABBR1 locus. The MOG gene (myelin oligodendrocyte glycoprotein) is a transmembrane protein expressed on the surface of myelin sheath, and may be involved in multiple sclerosis (OMIM#126200) [42, 43]; recent studies also found that MOG is differentially expressed in prefrontal cortex and temporal lobes in schizophrenic patients [43, 44]. The most significant interval (between SNPs rs1233399 and rs29225; top p = 3.4 × 10-3) spans the 3' end of the GABBR1 (Gamma-aminobutyric acid (GABA) B receptor, 1) and the overlapping UBD (Ubiquitin D) loci. GABBR1 is highly expressed in nervous systems and has been suggested as a candidate region for neuropsychiatric disorders [45, 46]. A recent study showed that the expression levels of GABBR1 are significantly lower in several brain regions in patients with autism compared with healthy controls [47]. Finally, we examined the median IBS distances between the subjects with regions of homozygosity on chromosomes 2 (13 Croatian cases and 10 Croatian controls) and chromosome 6 (17 Croatian cases and 19 Croatian controls) with permutation tests, but found that these subjects were not more closely related than the population average. Thus, the increased homozygosity in Croatia ASD cases cannot be explained by inbreeding among close relatives in a small number of selected families. However, the observed homozygosity may be the result of shared ancestral alleles from generations back in time.

UCSC Genome Browser genomic annotation around the region chr2:82,040,863-82,677,336. Genomic annotations of a region where ROH status is associated with case/control status in the Croatia cohort.

UCSC Genome Browser genomic annotation around the region chr6:29,481,192-29,719,410. Genomic annotations of a region where ROH status is associated with case/control status in the Croatia cohort.
Several studies have shown a role for copy number variation in predisposing individuals to complex diseases [48], including ASD [15, 49–52]. To address this, we next analyzed CNV calls from the high-density SNP genotyping data in Croatia cases and control subjects. We used PennCNV [53], a high-resolution CNV detection method, to call CNVs from the signal intensity data. We identified 2,303 CNVs (using a 10-SNP threshold) in 304 subjects passing the quality control criteria (Table 6). The median size of CNVs is 71.4 Kb, while the mean size of CNVs is 131.4 Kb. Due to the relatively small sample size, we do not expect that we could detect association of specific CNVs with disease status; instead, we focused on large and rare CNVs observed in cases but not controls, since they are more likely to be disease-related. For example, a recent study identified large (>500 Kb) CNVs to be especially pathogenic [54]; in addition, a CNV study on schizophrenia demonstrates that CNVs >500 Kb tend to be more prevalent in cases versus controls [55–57], and that CNVs >2 Mb are observed exclusively in cases [15, 16].
Examining 299 unrelated individuals passing QC, we identified a total of 32 CNVs >500 Kb, including 20 duplications and 12 deletions (Table 7 and Table 8). These large chromosomal rearrangements, detected in DNA samples isolated from blood (rather then cell lines), were confirmed through signal intensity data for the entire chromosome in the Illumina BeadStudio software (data not shown). Among them, 5/103 cases (4.85%) and 1/197 controls subject (0.49%) have CNVs greater than 2 Mb. An exceptionally large CNV was detected in an ASD female that harbored an 18 Mb duplication on chromosome 7 (7q21.11-21.3), which includes several known neurodevelopmental genes (PCLO, SEMA3A, SEMA3C, SEMA3D and SEMA3E). A duplication of this chromosomal region has been associated with intellectual disability and multiple congenital anomalies [58]. Another female has a 10 Mb deletion on chromosome 11 (11q24.2-q25). The Jacobsen syndrome (MIM147791) is a contiguous gene syndrome caused by terminal deletions of the long arm of chromosome 11 and typically associated with growth and psychomotor retardation and characteristic facial dimorphism [59].
Three of the ASD cases have multiple large rearrangements; one adult male harbors two chromosomal rearrangements (separated by 117 Mb) on chromosome 1, a 335 Kb deletion on 1p35.3 and a 882 Kb duplication on 1q21.1 (Figure 7a). In a second individual (adult female) we identified two distinct large CNVs: a 838 Kb deletion in an intergenic region between GRM8 and POT1 on 7q31.33 (chr7:124,752,465-125,591,197) and a 691 Kb deletion at 22q13.33 (chr22:48,833,840-49,524,956). This deletion encompasses the SHANK3 gene and cytogenetic rearrangements affecting this gene have been previously associated with ASD [60, 61]. It has been previously reported that the 22q13 deletion syndrome, also known as Phelan-McDermid syndrome (MIM606232), is characterized by severe neonatal hypotonia and global developmental delay, normal to accelerated growth, absent to severely delayed speech, and minor dysmorphic features [62, 63]. The third individual (male child) with multiple large rearrangements harbors a 534 Kb intergenic deletion at 2q21.1 (chr2: 129,716,352-130,250,751) and a 400 Kb duplication at 3q29 (chr3:195,987,870-196,387,903). These large structural variants, especially those found on multiple chromosomes illustrate the need to combine high-density genotype analysis with karyotype analysis to test for the presence of balanced translocations and inversions.

CNV status of AGRE, NINDS control, and Croatia ASD/control cohorts. (A) CNVs around four candidate genes (ACP6, CHD1L, FMO5, and PRKAB2) in 882 kb duplication on 1q21.1 (chr1:144,943,150-145,824,905). (B) Duplication of size 979 kb around the neuronal amiloride-sensitive cation channel gene (ACCN1) on Chr 17q11.2-q12 (chr17:28,000,000-30,100,000).
In contrast to large rearrangements encompassing >10 Mbs that were observed in ASD cases, the largest rearrangement observed in control subjects is a 2.5 Mb duplication at 15q24.1-q24.2 (chr15:70,783,089-73,316,235), a known microdeletion/duplication region [64]. Also, we found one control subject to have two duplications within the DiGeorge (MIM188400) and Velocardiofacial (MIM192430) syndrome region on chromosome 22 (22q11.21), a known pathogenic rearrangement hotspot where large (>1.5 Mb) deletions are pathogenic [65]. These duplications (922 Kb in chr22:17,690,812-18,613,429 and 563 Kb in chr22:19,102,598-19,665,559) are separated by 489 Kb. The observation that we see more large CNVs associated with ASD subjects (>2 Mb: 5 cases, 1 control; Fisher's test; p = 0.019) suggests these large rearrangements may be rare ASD susceptible variants; follow-up investigations on larger cohorts will be necessary.
Conclusions
In this paper, we report analysis of a genome-wide high-density genotype study for ASD in Croatia. The dataset, although small, provides a distinctive opportunity to evaluate structural variation associated with a complex disease such as ASD in a genetically distinct population. In addition to the analysis of ASD, high-density genotype data from a well-defined geographic area of Croatia permitted also the study of the subjects' genetic substructure and diversity. We asked whether the subjects included in this study which were collected throughout Croatia, show a lower level of genomic diversity in comparison to subjects from other defined demographic areas. Addressing this issue may suggest that a set of 100 Croatian ASD subjects harbor a smaller set of susceptibility loci in comparison a randomly selected set of ASD subjects from a larger area. The Croatian samples represent a genetic continuum of relatedness with a subset of individuals closer to those from several regions in Europe, probably due to numerous historic migrations. The impact of fine-scale population history on the genetic substructure of individuals from Croatia has recently been evaluated using a small set of selected loci [21]. To our knowledge, this study is the first to provide a genome-wide view of the genetic composition of the Croatia population.
Although our study represents the first high-resolution genomic analysis of subjects with ASD in Croatia, several caveats need to be addressed. Apart from an obviously limited sample size, the classification of ASD subjects in this pilot project has been established using the DSM-IV criteria. Due to phenotypic heterogeneity of ASD and our finding of a wide-range of novel, and in some cases, exceptionally large CNVs, there is a need for accurate clinical diagnostic criteria for the diagnosis of ASD and related neurodevelopmental syndromes. Currently no other diagnostic tools were available in Croatian; in order to compare our findings to those in other large-scale genetic studies it will be necessary to translate gold standard diagnostic tools such as ADOS [66] and ADI-R [67] to Croatian and many other languages.
We used PennCNV to call copy number variations, though other CNV calling programs such as QuantiSNP [68] and cnvPartition (a built-in software tool in the Illumina BeadStudio software)are available An advantage of using multiple CNV calling programs is to reduce false positives, namely one can focus on CNVs returned by multiple programs and expect them to be "more reliable". Since our study focused on large CNVs (>500 Kb) we were able to validate these variants by visual inspection of the raw intensities(data not shown).
We found that runs of homozygosity are more frequent in Croatia ASD subjects than either Croatia or NINDS control cohorts. Given the low frequency of subjects with ROH, larger follow-up studies are necessary to confirm the findings. Our findings suggest there is a high chance that homozygosity mapping based on family studies can discover rare recessive variants. Our finding that several ASD subjects harbor exceptionally large chromosomal rearrangements previously associated with distinct clinical syndromes such as Velocardiofacial, Jacobsen, Phelan-McDermid, or 3q29 microdeletion/duplication syndromes (MIM609425; MIM611936) lends support for cytogenetic or high-density genotype testing for ASD subjects with complex clinical manifestation [69]. Furthermore, recent findings that seemingly diverse neurodevelopmental and neurological disorders are often associated with the same chromosomal rearrangement(s) provide support for in-depth phenotype analysis and longitudinal clinical evaluation of entire families. These family-based studies may also reveal whether observed structural variants are de novo events or rather inherited and present in additional family members. We suggest that deeper population screens combined with family-based genetic analyses will lead to improved understanding of joint actions of associated loci and molecular basis of neurodevelopmental disorders in populations worldwide.
Methods
Subjects and DNA Preparation
The patient group consisted of 103 individuals (81 male, 22 female, mean age 21.5 ± 10.3, ranging from 4 to 45) recruited from the Centers for Autism in Zagreb, Rijeka and Split (Republic of Croatia) and diagnosed with autism spectrum disorders according to DSM-IV criteria. The control group consisted of 203 healthy blood donors (146 male, 57 female, mean age 32.5 ± 8.06, ranging from 19 to 45) [30] with no history of mental illnesses, behavioral disorders, or substance abuse. All subjects were of Croatian (southern Slavic) origin. After an informative talk, a written consent for inclusion in the study was obtained from the control subjects and from the patients' parents. The study has been carried out in accord with the Declaration of Helsinki, and was approved by the Ethics Committee of the Medical Faculty of the University of Zagreb.
Blood sampling was performed in the Centers for Autism (ASD cases) and in the Croatian Institute of Transfusion Medicine (control subjects) between 9 and 11 a.m. Either 2 or 5 ml of venous blood, depending on the age of the participants, was collected into vacutainers containing EDTA anticoagulant. DNA was isolated from the whole blood using a DNA isolation kit for mammalian blood (Boehringer Manheim, Germany).
Datasets and Preprocessing
The genomic DNA from blood was used to obtain genotypes by the Illumina HumanHap550 version 3 high-density array with 561,446 SNP markers. Coordinates of SNPs as well as genomic annotations are based on NCBI human genome build 36.3, (UCSC Genome Browser Release 18). The genotyping experiments were performed at the Center for Applied Genomics, Children's Hospital of Philadelphia, as previously described [70]. The raw genotyping signal data were processed by the Illumina BeadStudio software and converted to normalized signal intensity values, represented as Log R Ratio (LRR) and B Allele Frequency (BAF). Due to the presence of "genomic wave patterns" in some of the genotyped samples, we applied a data pre-processing protocol [71] to increase the signal-to-noise ratio of the LRR values for all samples.
We analyzed three genome-wide datasets: Croatia Autism cohort (306 subjects), NINDS Control cohort (542 subjects), and genotyping data from the Human Genetic Diversity Panel (1043 subjects, genotyped at Stanford University, downloaded from http://hagsc.org/hgdp/files.html). The quality control (QC) procedure (described in detail in Wang et al. [14] and Bucan et al. [16]), done separately for the three datasets, is as follows: (1) we first discarded SNPs with 5% or more missing genotyping calls; (2) we then recomputed summary statistics for individuals and markers; (3) we retained individuals/markers passing the following threshold: individual - missing genotyping call percentage ≤5%; marker - missing genotyping call percentage ≤5%, minor allele frequency ≥5%. We then merged the Croatia cohort with the NINDS control and HGDP datasets separately into two combined datasets (Croatia samples and HGDP, Croatia samples and NINDS), including all individuals passing QC in their separate datasets, and retaining only markers passing QC in both datasets. Subpopulation membership for HGDP samples were obtained from Supplemental material from Kidd et al. [72]
To detect hidden related samples, we computed , the estimated probability of IBD between every pair of subjects from the Croatia control and case and NINDS control samples using PLINK. We found 5 pairs of Croatia control samples (but none among the case samples) and 4 pairs of NINDS control samples have > 0.25 (siblings or closer). To be more specific, three Croatia control pairs and one NINDS control pair have > 0.999 (duplicate genotyping or monozygotic twins), and two Croatia Ctrl pairs and three NINDS control pairs have around 0.5 (first-degree relatives). To reduce relatedness, in subsequent analysis, for each pair we removed the one of the two samples with a larger index number in the ROH analysis. Table 1 summarizes the three genome-wide datasets analyzed in this paper.
IBS distance analysis
We computed the IBS (identity by state) distance between any two subjects using PLINK, and constructed three pairwise distance matrices: Croatia controls and HGDP, Croatia controls and NINDS, Croatia cases and controls. The IBS distance between two subjects is defined as A+B/2, where A and B are the number of SNPs that differ by one and two alleles between the two subjects, respectively. We performed multidimensional scaling using R [73] and plotted the first two coordinates, and built trees using FastME, a distance-based phylogeny reconstruction software [74].
FST Analysis
To quantitatively describe the genetic difference between two population we next used the FST coefficient (fixation coefficient). This measure combines information across many loci in many individuals and a higher FST value for a locus indicates difference between the genetic compositions in two populations, whereas FST = 0 implies no discernable difference between two populations at the locus.
We analyzed the FST values using the Croatia control and NINDS control cohorts as follows. We first computed the heterozygosity of each marker (defined as the proportion of heterozygous individuals in the population) using PLINK, then computed the fixation index F ST from the heterozygosity by R by the following steps. (1) Compute the minor allele frequencies p1, p2 and p for the two subpopulations (with N1 and N2 subjects) and the whole population (with N = N1 + N2 subjects). (2) Compute the expected proportion of heterozygotes HT = p (1-p) assuming the two subpopulations are genetically identical for the marker. (2) Compute the expected proportion of heterozygotes HS = (N1 p1 (1-p1) + N2 p2 (1-p2))/N. (3) Compute .
Runs of homozygosity
Runs of homozygosity (ROHs) are extended genomic regions where the DNA sequences on the two chromosomes are identical, and may be due to identity by descent or gene conversion; since polymorphic sites are all homozygous, SNP arrays can be used to identify such regions. Study of ROHs can provide information on the population structure such as extent of inbreeding, and can be used to find candidate genes in recessive disorders. We computed the distribution of runs of homozygosity for three groups (Croatia case, control, NINDS) using PLINK with the following settings: an interval passes as an ROH if it has 100 or more homozygous SNPs (except at most one SNP), with density at least one SNP every 50 Kb and at most 1 Kb gap between any two neighboring SNPs. We ran Wilcoxon test between every pair of the three groups, as well as Kruskal-Wallis test for comparing three groups simultaneously.
To identify genomic regions where the ROH status correlates with the case/control status, we ran Fisher's test as follows: we first sorted the 7,766 endpoints of all autosomal ROHs across Croatia case and control groups; for each pair of adjacent endpoints, we computed the significance of association between ROH status and case/control status by Fisher's test.
Correlation between ROH and age
Motivated by the study demonstrating the positive correlation between runs of homozygosity and age in a North American cohort of European descent due to urbanization [35], we examined the correlation between age and ROH frequency and size. Only the 103 ASD subjects have age information. We used age and sex as covariates in a full linear regression model (in R/S-Plus specification):
Here the dependent variable can be either the total ROH length, average ROH length, or number of ROH regions in a subject.
CNVs analysis
A previously described high-resolution CNV detection algorithm, the PennCNV algorithm [53], was used to infer CNVs from the signal intensity data following protocols described in Wang et al. [53] The PennCNV algorithm uses a Hidden Markov model to identify any genomic regions with duplications or deletions by combining signal intensity and SNP allelic ratio distributions. The Illumina BeadStudio software provided visualization tools that were used to display signal intensity data for the entire chromosome.
References
Abrahams BS, Geschwind DH: Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008, 9 (5): 341-355. 10.1038/nrg2346.
Sadock BJ, Kaplan HI, Sadock VA: Kaplan & Sadock's synopsis of psychiatry : behavioral sciences/clinical psychiatry. 2007, Philadelphia: Wolter Kluwer/Lippincott Williams & Wilkins, 10
Muhle R, Trentacoste SV, Rapin I: The genetics of autism. Pediatrics. 2004, 113 (5): e472-486. 10.1542/peds.113.5.e472.
Lockyer L, Rutter M: A five- to fifteen-year follow-up study of infantile psychosis. Br J Psychiatry. 1969, 115 (525): 865-882. 10.1192/bjp.115.525.865.
Rutter M: Autistic children: infancy to adulthood. Semin Psychiatry. 1970, 2 (4): 435-450.
Volkmar FR, Lord C, Bailey A, Schultz RT, Klin A: Autism and pervasive developmental disoders. Journal of Child Psychology and Psychiatry. 2004, 45: 135-170. 10.1046/j.0021-9630.2003.00317.x.
Rutter M, Silberg J, O'Connor T, Simonoff E: Genetics and child psychiatry: II Empirical research findings. J Child Psychol Psychiatry. 1999, 40 (1): 19-55. 10.1111/1469-7610.00423.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M: Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995, 25 (1): 63-77. 10.1017/S0033291700028099.
Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al: Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007, 39 (3): 319-328. 10.1038/ng1985.
Gupta AR, State MW: Recent advances in the genetics of autism. Biol Psychiatry. 2007, 61 (4): 429-437. 10.1016/j.biopsych.2006.06.020.
Vorstman JA, Staal WG, van Daalen E, van Engeland H, Hochstenbach PF, Franke L: Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006, 11 (1): 18-28. 10.1038/sj.mp.4001757. 1
Freitag CM: The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007, 12 (1): 2-22. 10.1038/sj.mp.4001896.
Veenstra-VanderWeele J, Cook EH: Molecular genetics of autism spectrum disorder. Mol Psychiatry. 2004, 9 (9): 819-832. 10.1038/sj.mp.4001505.
Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al: Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009, 459 (7246): 528-533. 10.1038/nature07999.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al: Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009, 459 (7246): 569-573. 10.1038/nature07953.
Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, et al: Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009, 5 (6): e1000536-10.1371/journal.pgen.1000536.
Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al: Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008, 321 (5886): 218-223. 10.1126/science.1157657.
Peltonen L, Palotie A, Lange K: Use of population isolates for mapping complex traits. Nat Rev Genet. 2000, 1 (3): 182-190. 10.1038/35042049.
Kristiansson K, Naukkarinen J, Peltonen L: Isolated populations and complex disease gene identification. Genome Biol. 2008, 9 (8): 109-10.1186/gb-2008-9-8-109.
Darvasi A, Shifman S: The beauty of admixture. Nat Genet. 2005, 37 (2): 118-119. 10.1038/ng0205-118.
Rudan I, Rudan D, Campbell H, Carothers A, Wright A, Smolej-Narancic N, Janicijevic B, Jin L, Chakraborty R, Deka R, et al: Inbreeding and risk of late onset complex disease. J Med Genet. 2003, 40 (12): 925-932. 10.1136/jmg.40.12.925.
McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, et al: Runs of homozygosity in European populations. Am J Hum Genet. 2008, 83 (3): 359-372. 10.1016/j.ajhg.2008.08.007.
Jakobsson M, Scholz SW, Scheet P, Gibbs JR, VanLiere JM, Fung HC, Szpiech ZA, Degnan JH, Wang K, Guerreiro R, et al: Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008, 451 (7181): 998-1003. 10.1038/nature06742.
Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008, 319 (5866): 1100-1104. 10.1126/science.1153717.
Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al: Genes mirror geography within Europe. Nature. 2008, 456 (7218): 98-101. 10.1038/nature07331.
Hranilovic D, Bujas-Petkovic Z, Vragovic R, Vuk T, Hock K, Jernej B: Hyperserotonemia in adults with autistic disorder. J Autism Dev Disord. 2007, 37 (10): 1934-1940. 10.1007/s10803-006-0324-6.
Hranilovic D, Novak R, Babic M, Novokmet M, Bujas-Petkovic Z, Jernej B: Hyperserotonemia in autism: the potential role of 5HT-related gene variants. Coll Antropol. 2008, 32 (Suppl 1): 75-80.
Hranilovic D, Bujas-Petkovic Z, Tomicic M, Bordukalo-Niksic T, Blazevic S, Cicin-Sain L: Hyperserotonemia in autism: activity of 5HT-associated platelet proteins. J Neural Transm. 2009, 116 (4): 493-501. 10.1007/s00702-009-0192-2.
American Psychiatric Association, American Psychiatric Association, Task Force on DSM-IV: Diagnostic and statistical manual of mental disorders : DSM-IV-TR. 2000, Washington, DC: American Psychiatric Association, 4
Schopler E, Reichler RJ, DeVellis RF, Daly K: Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J Autism Dev Disord. 1980, 10 (1): 91-103. 10.1007/BF02408436.
Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, et al: ASPM is a major determinant of cerebral cortical size. Nat Genet. 2002, 32 (2): 316-320. 10.1038/ng995.
Piao X, Hill RS, Bodell A, Chang BS, Basel-Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM, et al: G protein-coupled receptor-dependent development of human frontal cortex. Science. 2004, 303 (5666): 2033-2036. 10.1126/science.1092780.
Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, et al: Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004, 75 (6): 979-987. 10.1086/425985.
Lencz T, Lambert C, DeRosse P, Burdick KE, Morgan TV, Kane JM, Kucherlapati R, Malhotra AK: Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc Natl Acad Sci USA. 2007, 104 (50): 19942-19947. 10.1073/pnas.0710021104.
Nalls MA, Simon-Sanchez J, Gibbs JR, Paisan-Ruiz C, Bras JT, Tanaka T, Matarin M, Scholz S, Weitz C, Harris TB, et al: Measures of autozygosity in decline: globalization, urbanization, and its implications for medical genetics. PLoS Genet. 2009, 5 (3): e1000415-10.1371/journal.pgen.1000415.
Simon-Sanchez J, Scholz S, Fung HC, Matarin M, Hernandez D, Gibbs JR, Britton A, de Vrieze FW, Peckham E, Gwinn-Hardy K, et al: Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet. 2007, 16 (1): 1-14. 10.1093/hmg/ddl436.
Gibbs JR, Singleton A: Application of genome-wide single nucleotide polymorphism typing: simple association and beyond. PLoS Genet. 2006, 2 (10): e150-10.1371/journal.pgen.0020150.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81 (3): 559-575. 10.1086/519795.
Cornish KM, McManus IC: Hand preference and hand skill in children with autism. J Autism Dev Disord. 1996, 26 (6): 597-609. 10.1007/BF02172349.
Francks C, Maegawa S, Lauren J, Abrahams BS, Velayos-Baeza A, Medland SE, Colella S, Groszer M, McAuley EZ, Caffrey TM, et al: LRRTM1 on chromosome 2p12 is a maternally suppressed gene that is associated paternally with handedness and schizophrenia. Mol Psychiatry. 2007, 12 (12): 1129-1139. 10.1038/sj.mp.4002053. 1057
Ludwig KU, Mattheisen M, Muhleisen TW, Roeske D, Schmal C, Breuer R, Schulte-Korne G, Muller-Myhsok B, Nothen MM, Hoffmann P, et al: Supporting evidence for LRRTM1 imprinting effects in schizophrenia. Mol Psychiatry. 2009, 14 (8): 743-745. 10.1038/mp.2009.28.
Boyle LH, Traherne JA, Plotnek G, Ward R, Trowsdale J: Splice variation in the cytoplasmic domains of myelin oligodendrocyte glycoprotein affects its cellular localisation and transport. J Neurochem. 2007, 102 (6): 1853-1862. 10.1111/j.1471-4159.2007.04687.x.
Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Maccarrone G, Dias-Neto E, Turck CW: Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2009, 259 (3): 151-163. 10.1007/s00406-008-0847-2.
Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Marangoni S, Novello JC, Maccarrone G, Turck CW, Dias-Neto E: Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J Neural Transm. 2009, 116 (3): 275-289. 10.1007/s00702-008-0156-y.
Burfoot RK, Jensen CJ, Field J, Stankovich J, Varney MD, Johnson LJ, Butzkueven H, Booth D, Bahlo M, Tait BD, et al: SNP mapping and candidate gene sequencing in the class I region of the HLA complex: searching for multiple sclerosis susceptibility genes in Tasmanians. Tissue Antigens. 2008, 71 (1): 42-50.
Andrieux J, Richebourg S, Duban-Bedu B, Petit F, Lepretre F, Sukno S, Dehouck MB, Delobel B: Characterization by array-CGH of an interstitial de novo tandem 6p21.2p22.1 duplication in a boy with epilepsy and developmental delay. Eur J Med Genet. 2008, 51 (4): 373-381. 10.1016/j.ejmg.2008.02.010.
Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD: Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009, 8 (1): 64-69. 10.1007/s12311-008-0075-3.
Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet. 2006, 7 (2): 85-97. 10.1038/nrg1767.
Maestrini E, Pagnamenta AT, Lamb JA, Bacchelli E, Sykes NH, Sousa I, Toma C, Barnby G, Butler H, Winchester L, et al: High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol Psychiatry. 2009, 15: 954-968. 10.1038/mp.2009.34.
Sykes NH, Toma C, Wilson N, Volpi EV, Sousa I, Pagnamenta AT, Tancredi R, Battaglia A, Maestrini E, Bailey AJ, et al: Copy number variation and association analysis of SHANK3 as a candidate gene for autism in the IMGSAC collection. Eur J Hum Genet. 2009, 17 (10): 1347-1353. 10.1038/ejhg.2009.47.
Noor A, Gianakopoulos PJ, Fernandez B, Marshall CR, Szatmari P, Roberts W, Scherer SW, Vincent JB: Copy number variation analysis and sequencing of the X-linked mental retardation gene TSPAN7/TM4SF2 in patients with autism spectrum disorder. Psychiatr Genet. 2009, 19 (3): 154-155. 10.1097/YPG.0b013e32832a4fe5.
Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, et al: Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010, 466: 368-372. 10.1038/nature09146.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M: PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17 (11): 1665-1674. 10.1101/gr.6861907.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, et al: Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009, 84 (2): 148-161. 10.1016/j.ajhg.2008.12.014.
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, et al: Large recurrent microdeletions associated with schizophrenia. Nature. 2008, 455 (7210): 232-236. 10.1038/nature07229.
Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, Nelson SF: Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet. 2004, 75 (6): 1117-1123. 10.1086/426034.
Walsh CA, Morrow EM, Rubenstein JL: Autism and brain development. Cell. 2008, 135 (3): 396-400. 10.1016/j.cell.2008.10.015.
Lukusa T, Fryns JP: Syndrome of facial, oral, and digital anomalies due to 7q21.2-->q22.1 duplication. Am J Med Genet. 1998, 80 (5): 454-458. 10.1002/(SICI)1096-8628(19981228)80:5<454::AID-AJMG4>3.0.CO;2-O.
Mattina T, Perrotta CS, Grossfeld P: Jacobsen syndrome. Orphanet J Rare Dis. 2009, 4: 9-10.1186/1750-1172-4-9.
Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H, et al: Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007, 39 (1): 25-27. 10.1038/ng1933.
Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, Zwaigenbaum L, Fernandez B, Roberts W, Szatmari P, et al: Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007, 81 (6): 1289-1297. 10.1086/522590.
Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE: Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003, 40 (8): 575-584. 10.1136/jmg.40.8.575.
Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O: Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001, 69 (2): 261-268. 10.1086/321293.
Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al: Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006, 38 (9): 1038-1042. 10.1038/ng1862.
Emanuel BS, Saitta SC: From microscopes to microarrays: dissecting recurrent chromosomal rearrangements. Nat Rev Genet. 2007, 8 (11): 869-883. 10.1038/nrg2136.
Lord C, Rutter M, Goode S, Heemsbergen J, Jordan H, Mawhood L, Schopler E: Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J Autism Dev Disord. 1989, 19 (2): 185-212. 10.1007/BF02211841.
Lord C, Rutter M, Le Couteur A: Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994, 24 (5): 659-685. 10.1007/BF02172145.
Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J: QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35 (6): 2013-2025. 10.1093/nar/gkm076.
Lintas C, Persico AM: Autistic phenotypes and genetic testing: state-of-the-art for the clinical geneticist. J Med Genet. 2009, 46 (1): 1-8. 10.1136/jmg.2008.060871.
Hakonarson H, Grant SF, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Casalunovo T, Taback SP, Frackelton EC, et al: A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007, 448 (7153): 591-594. 10.1038/nature06010.
Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, et al: Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009, 459 (7249): 987-991. 10.1038/nature08035.
Kidd JM, Newman TL, Tuzun E, Kaul R, Eichler EE: Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007, 3 (4): e63-10.1371/journal.pgen.0030063.
R Development Core Team: R: A language and environment for statistical computing. 2005, Vienna, Austria: R Foundation for Statistical Computing
Desper R, Gascuel O: Getting a tree fast: Neighbor Joining, FastME, and distance-based methods. Curr Protoc Bioinformatics. 2006, Chapter 6 (Unit 6): 3-
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/11/134/prepub
Acknowledgements
This study was supported by grant from the Ministry of Science Education and Sports of the Republic of Croatia (grants # 119-1081870-2396 and 098-1081870-2395) and a Penn Genome Frontiers Institute internal grant (LW). Genome-wide genotyping of the Croatian cohort was funded by an Institutional Development Award to the Center for Applied Genomics from the Children's Hospital of Philadelphia. Andrew Singleton (NIH) kindly granted us access to the NINDS control dataset. We thank Andrew Singleton and Nancy Spinner (University of Pennsylvania) for their insightful comments on our manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MB, DH and HH designed the study. DH, ZBP, and BJ contributed samples. LW, KW, IL, LY, NG, HH and MB participated in the analysis. LW and MB wrote the paper. All co-authors contributed to the preparation of the paper.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, LS., Hranilovic, D., Wang, K. et al. Population-based study of genetic variation in individuals with autism spectrum disorders from Croatia. BMC Med Genet 11, 134 (2010). https://doi.org/10.1186/1471-2350-11-134
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-11-134