
Abstract
Background
The Human Immunodeficiency Virus type 1 (HIV-1) envelope glycoprotein gp160, useful in detecting anti-HIV-1 antibodies, is difficult to express in heterologous hosts. The major hurdles are its signal sequence, strong hydrophobic regions and heavy glycosylation. While it has not been possible to express full length recombinant (r)-gp160 in E. coli, it can be expressed in insect and mammalian cells, but at relatively higher cost. In this work, we report E. coli-based over-expression of r-gp160 variant and evaluate its performance in diagnostic immunoassays for the detection of anti-HIV-1 antibodies.
Methods
A deletion variant of r-gp160 lacking hydrophobic regions of the parent full length molecule was expressed in E. coli and purified to near homogeneity using single-step Ni(II)-affinity chromatography. Biotinylated and europium(III) chelate-labeled versions of this antigen were used to set up one- and two-step time-resolved fluorometric double antigen sandwich assays. The performance of these assays was evaluated against a collection of well-characterized human sera (n=131), that included an in-house panel and four commercially procured panels.
Results
In-frame deletion of three hydrophobic regions, spanning amino acid residues 1–43, 519–538 and 676–706, of full length HIV-1 gp160 resulted in its expression in E. coli. Both the one- and two-step assays manifested high sensitivity unambiguously identifying 75/77 and 77/77 HIV-1 positive sera, respectively. Both assays also identified all 52 HIV-seronegative sera correctly. Between the two assays, the mean signal-to-cutoff value of the two-step assay was an order of magnitude greater than that of the one-step assay. Both assays were highly specific manifesting no cross-reactivity towards antibodies specific to other viruses like hepatitis B, C, and human T cell leukemia viruses.
Conclusions
This study has demonstrated the expression of r-gp160 variant in E. coli, by deletion of hydrophobic regions, and its purification in reasonable yields. This underscores the potential for cost saving in antigen production. Evaluation of this antigen in a double antigen sandwich two-step assay showed it to be a highly sensitive and specific HIV-1 diagnostic reagent. The amenability of this assay to the one-step format suggests its potential utility in developing a rapid point-of-care HIV-1 diagnostic test.
Similar content being viewed by others

Background
Human immunodeficiency virus (HIV), a member of the family Retroviridae, is the causative agent of acquired immunodeficiency syndrome (AIDS), characterized by profound immune system dysfunction [1]. Recent estimates of the global AIDS burden indicate that there were 2.7 million new infections (bringing the total number to ~34 million infected individuals worldwide) and 1.8 million deaths in 2010 [2]. Diagnostic tests play a critical role in curtailing the spread of HIV, through early detection of infection to identify individuals needing antiviral therapy, and in screening blood and blood related products [3–5]. Most commonly used HIV screening tests are based on the principle of enzyme immunoassay (EIA) that detect either the antibodies (first, second and third generation EIAs) or p24 antigen as well (fourth generation EIA) [6, 7]. The envelope glycoprotein (gp160) of HIV-1 is well documented as a viral antigen that presents immunodominant regions for serodiagnosis of HIV-1 infections [8–11]. Native gp160 which can be obtained by HIV-1 infection of H9 cells in tissue culture [12] has been shown to detect earliest HIV-1 seroconversion in different EIAs [12, 13]. The inherent biohazard entailed in gp160 production can be circumvented through recombinant expression. However, the gp160 antigen, with its strong hydrophobic regions and heavy glycosylation, is a difficult candidate for heterologous expression and purification [8, 14, 15]. Nevertheless, recombinant (r)-gp160 can be expressed in insect and mammalian cells, and purified by immunoaffinity or lectin affinity chromatography [14–18]. Though both native and r-gp160 are commercially available, the expensive expression and purification systems increase the production cost of the protein, and in turn contribute to increased cost of HIV testing.
One way to cut down the cost of HIV testing would be to try and exploit the high production potential of prokaryotic expression hosts to produce r-gp160. However, full-length r-gp160 expression in E. coli has not been successful. It was reported that a plasmid containing full-length HIV-1 gp160 insert was toxic to E. coli resulting in either elimination or rearrangement of the insert [19]. It has been reported that there are two hydrophobic regions in the C-terminal half of gp160 [8]. In this study, we deleted these two regions, as well as the hydrophobic signal peptide, implicated in low expression in mammalian cells [14, 15], to create a deletion mutant, Δgp160, and found this to be expressed quite well in E. coli, leading to the purification of 15–20 mg r-Δgp160 antigen per liter of induced culture. Compared to the native full-length gp160, the r-Δgp160 antigen developed in this study is almost full-length, lacking just ~10% of the parent sequences (representing three hydrophobic regions). We used two versions of this antigen to set up one- and two-step time-resolved fluorometric (TRF) double antigen sandwich (bridge) assays as described earlier [20]. One version of the antigen was biotinylated [immobilized on streptavidin (SA)-coated microtiter wells, to capture anti-HIV antibodies] and the other, labeled with a europium (Eu3+) chelate (as a tracer to reveal bound antibodies). By testing a collection of well-characterized human sera (n=131) in these assays, we provide evidence to show that the E.coli-expressed r-Δgp160 antigen is a potential diagnostic tool of high sensitivity and specificity for the detection of anti-HIV-1 antibodies.
Methods
Materials
E. coli host strains DH5α and BL21(DE3), used for cloning and expression, respectively, were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). Plasmid pET-28a(+) was from Novagen (Madison, WI, USA). Plasmids pTrxBAP and pBirA have been described before [20, 21]. Enzymes for routine cloning work were procured from MBI Fermentas (Burlington, Canada). Ni-NTA super flow resin and isopropyl-β-D-thiogalactopyranoside (IPTG) were from Qiagen (Hilden, Germany), and Calbiochem-EMD Biosciences (La Jolla, CA, USA), respectively. The intrinsically fluorescent isothiocyanate activated nonadentate Eu3+-chelate [22, 23] and biotin isothiocyanate [24] for antigen labeling were synthesized as before. Nucleic Acid Purification (NAP) Sephadex G-25 columns were procured from GE Healthcare (Uppsala, Sweden). SA-coated normal capacity low-fluorescent microtiter plates were obtained from Kaivogen Oy (Turku, Finland).
A collection of 131 human sera samples was assembled for the study. This consisted of one in-house panel and 4 commercially procured panels. The in-house panel of 59 sera samples (consisting of 22 HIV positive and 37 HIV negative sera), pre-screened using Vidas HIV Duo Quick kit (bioMérieux SA, Marcy I’Etoile, France), was obtained from the Department of Virology, University of Turku. These sera samples were collected as per the guidelines of Ethics Committee of the Turku University Hospital. Well-characterized worldwide HIV performance panel (WWRB 302–01 to WWRB 302–30), HIV-1 seroconversion panel (PRB 931–01 to PRB 931–09), anti-HIV-1 low titer performance panel (PRB 108–1 to PRB 108–15) and viral co-infection panel (PCA 201–01 to PCA 201–25) were purchased from SeraCare Life Sciences (Milford, MA, USA). In total, there were 77 anti-HIV-1 positive, 2 anti-HIV-2 positive and 52 anti-HIV negative samples.
Generation of recombinant gp160 antigens
The 760 amino acid (aa) residue long r-Δgp160 antigen was designed by eliminating the signal sequence (aa 1–43) and two internal hydrophobic regions (aa 519–538 and 676–706) of HIV-1 (strain NL4-3). The corresponding synthetic gene, codon-optimized for E. coli expression was obtained from Geneart (Regensburg, Germany), and used to express two variants of the encoded recombinant antigen. This synthetic gene was inserted into (i) pET-28a(+), in-frame with the vector-encoded 6x-His tag and stop codon at its carboxyl end, to express r-Δgp160 antigen, and (ii) pTrxBAP in frame with the vector-encoded Trx (thioredoxin)-6x-His tag-BAP sequences at the amino terminus, to express the r-Trx-BAP-Δgp160 antigen. The resultant plasmids were introduced separately into E. coli BL21(DE3), and induced to express with 1 mM IPTG. In order to achieve biotinylation of the r-Trx-BAP-Δgp160 antigen in vivo, E. coli cells expressing this antigen were co-transformed with IPTG-inducible biotin ligase expressing plasmid pBirA and cultured in medium supplemented with biotin (10 μg/ml).
As both recombinant protein variants were associated with the insoluble fraction, they were purified from induced cells under denaturing conditions [20]. Briefly, induced cell pellets were re-suspended in lysis buffer (6 M guanidine HCl/20 mM Tris–HCl/300 mM NaCl/10% glycerol/0.1% sodium deoxycholate/1% Tween-20/10 mM β-mercaptoethanol/20 mM imidazole, pH 8) and lysed by sonication at 4°C (Sonics Vibracell sonicator). The resulting lysate was clarified and chromatographed on a 5 ml Ni-NTA super flow resin column. After washing the column (wash buffer was similar to lysis buffer lacking sodium deoxycholate with the exception that 6 M Guanidine-HCl was replaced by 8 M urea), elution was performed using a linear imidazole gradient (20–500 mM) in wash buffer. All the eluted fractions were analyzed by SDS–PAGE, the peak fractions were pooled together, filtered through 0.22 μm membrane and stored in 1 ml aliquots at a concentration of about 1 mg/ml at −20°C until further use. One aliquot was thawed at a time, heat-treated at 80°C for 90 seconds, snap-cooled in ice bath and the buffer was changed to 50 mM sodium carbonate buffer, pH 9.8 using NAP-5 or NAP-10 column, for subsequent labeling reaction.
Preparation of labeled antigens
The r-Δgp160 antigen was labeled with a 30-fold molar excess of Eu3+-chelate at 4°C for 16–20 hours in 50 mM sodium carbonate buffer, pH 9.8, essentially as described before [20]. The r-Trx-BAP-Δgp160 (which failed to be efficiently biotinylated in vivo) was in vitro (chemically) biotinylated with a 40-fold molar excess of biotin isothiocyanate. The reaction was performed in 50 mM sodium carbonate buffer, pH 9.8, at room temperature (RT) for 4 hours. Both the unincorporated labels were removed by passing the reaction mixtures sequentially through NAP-5 and NAP-10 columns. For the sake of simplicity, Eu3+-chelate labeled r-Δgp160 and chemically biotinylated r-Trx-BAP-Δgp160 antigens are referred to as r-Δgp160-Eu3+ and r-Δgp160-Bio, respectively. The labeled antigens were supplemented with 0.1% bovine serum albumin and 0.05% sodium azide, filtered through 0.22 μm membranes and stored at 4°C, until further use.
In-house TRF double antigen sandwich assays
Either r-Δgp160-Bio antigen (600 ng/100 μl assay buffer/well) or the previously reported [20]in vivo biotinylated r-Bio-HIV-1env antigen (200 ng/100 μl assay buffer/well) was immobilized on SA-coated microtiter wells. The assay buffer was 50 mM sodium carbonate, pH 9.6 (containing 25 mM NaCl/0.1% Tween-20/2.5% BSA/1.25% sucrose/0.06% γ-globulins from bovine blood/0.05% NaN3). Binding of biotinylated antigen to SA-coated microtiter wells was allowed to proceed for 1 hour at RT with shaking. The wells were then rinsed twice with wash buffer (154 mM NaCl/0.5 M KCl/0.1% Tween-20/50 mM potassium phosphate buffer, pH 7.2). These antigen-coated wells were used in one- and two-step assay formats as described below.
In the one-step assay (performed only with r-Δgp160-Bio antigen-coated well), a mixture of 50 μl serum, diluted (1:10) in assay buffer, plus 50 μl assay buffer containing 100 ng r-Δgp160-Eu3+, was added to each well and incubated for 1 hour at RT with shaking. The wells were washed 7 times with wash buffer. Time-resolved fluorescence for Eu3+-chelate was measured (λex:340nm; λem:615nm) from dry wells using Victor3V 1420 Multilabel counter (modified standard europium protocol; measurement height 5 mm).
In the two-step assay, 100 μl serum (diluted 1:20 in assay buffer) was added into each well and incubated for 1 hour at RT with shaking. Following 4 washes, 100 μl assay buffer containing either 100 ng r-Δgp160-Eu3+ (where r-Δgp160-Bio was immobilized as capture antigen) or r-HIV-1env-Eu3+ (where r-Bio-HIV-1env was immobilized as capture antigen) was added to each well and incubated for 1 hour at RT with shaking. The wells were washed 7 times with wash buffer and read as above.
Data analysis
Each assay was evaluated with all the sera on the same day and the results obtained from different assays in single analysis were compared. Sera were designated as either positive or negative using cutoff values of 1000 counts per second (cps) and 924 cps, respectively for the one- and two-step r-Δgp160-based assays developed in this study. A cutoff value of 818 cps was used for the two-step r-HIV-1env assay, based on the previous study [20]. These cutoff values were obtained by adding 3 times the standard deviation (SD) to the mean read-out of 37 HIV-negative sera (of the in-house panel) in the one-step r-Δgp160- (mean = 598 cps; SD = 134 cps), two-step r-Δgp160- (mean = 509 cps; SD = 138 cps), and two-step r-HIV-1env- (mean = 577 cps; SD = 80 cps) antigen based assays described above. The use of three times SD to determine the cutoff value was based on a preliminary characterization of the assay with seronegative samples (n=37). Serum samples with ‘signal-to-cutoff’ (S/Co) ratios of <1.0 were designated as negative, while those with S/Co≥1.0 were designated as positive. Statistical significance of differences in sensitivities between assays was assessed using two-tailed Student’s t test. Differences were considered to be of statistical significance when the probability (p) was <0.05.
Results and discussion
Recombinant gp160 antigens
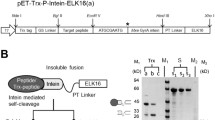
The two HIV-1 gp160 antigen variants, r-Δgp160 and r-Trx-BAP-Δgp160, created for this study are shown schematically in Figure 1. Both were designed to delete the amino-terminal signal peptide and two internal hydrophobic stretches of the native HIV-1 gp160 molecule without disruption of the reading frame. Additional sequences were added to these through the use of distinct expression vectors. Both were provided with 6x-His tags to facilitate purification. The key difference between the two was that the r-Trx-BAP-Δgp160 variant also contained at its amino terminus a thioredoxin (Trx)-biotin acceptor peptide (BAP) fusion partner. The purpose of the Trx moiety was to promote solubility of the recombinant antigen and thus facilitate its in vivo biotinylation at the target site provided by BAP. The two antigens were expressed by IPTG induction of BL21(DE3) host cells harboring the cognate expression constructs. In the case of r-Trx-BAP-Δgp160, bacterial biotin ligase was co-expressed to mediate biotinylation of its BAP moiety. An SDS-PAGE analysis of aliquots of cells before and after induction of expression of the r-Δgp160 and r-Trx-BAP-Δgp160 constructs is shown in Figure 2. It was difficult to discern the presence of a distinct induced polypeptide of the predicted molecular weights in both cases, though an additional band of ~34 kDa, consistent with the size of biotin ligase, was clearly seen in cells harboring the r-Trx-BAP-Δgp160 construct. Immunoblot analyses of these induced lysates with a monoclonal antibody specific to the 6x-His tag revealed that both r-Δgp160 and r-Trx-BAP-Δgp160 antigens are indeed present in the induced lysates. However, both recombinant antigens were found to be associated with the insoluble fraction (data not shown). Apparently, the Trx fusion is ineffective in promoting solubility of r-Δgp160 antigen. This is consistent with the previous observations that E. coli-expressed Trx fusion antigens are not necessarily soluble [20, 21]. Therefore, we attempted to purify these antigens under denaturing conditions. This resulted in near homogenous preparations of the two antigens, as evidenced by the SDS-PAGE profiles of the pooled peak fractions in Figure 2 (lanes marked ‘P’). The observed mobilities of purified r-Δgp160 and r-Trx-BAP-Δgp160 were consistent with their predicted sizes of ~88 and ~109 kDa, respectively. Our data show that deletion of the hydrophobic regions is compatible with the expression of r-Δgp160 variants in E. coli. Starting from 1L induced E. coli cultures we obtained ~20 and ~15 mg, respectively, of purified r-Δgp160 and r-Trx-BAP-Δgp160. However, as purifications were carried out in the presence of urea, it had to be removed from the purified antigens prior to their labeling for use in the double antigen sandwich assays (below). We did not determine if the purified proteins are correctly folded and retained the integrity of their conformational epitopes. However, the recognition of linear epitopes by their cognate antibodies may be sufficient to develop a sensitive immunoassay. Further, the hydrophobic regions deleted in r-Δgp160 are not exposed in the native structure of the full-length protein and therefore not expected to contribute to immunoreactivity [25]. It is relevant to point out that expression of full-length HIV-1 gp160 in E. coli has not been possible thus far due to instability/toxicity issues related to full-length, in-frame gp160 gene [19]. In mammalian cells, gp160 undergoes proteolysis in the Golgi complex to produce gp120 and gp41. It was shown recently that when gp120 is expressed in the absence of gp41 in mammalian cells it forms aberrant disulfide linked dimers [26]. Further, it has been reported that gp41 which can be expressed in E. coli, is associated with inclusion bodies regardless of whether it has a GST fusion partner [27, 28]. Purification under denaturing conditions was reported to yield ~5 mg [28] to ~12 mg [27] soluble protein/L of bacterial cell culture.

Design of r-Δgp160 and r-Trx-BAP-Δgp160 antigens and TRF assay. The left part of the figure shows a schematic representation of the native HIV-1 gp160 molecule. The black and grey boxes denote the signal sequence and the two internal hydrophobic regions in the C-terminal half of native gp160, respectively. Depicted in the middle part of the figure are the two recombinant antigens created for this study, r-Δgp160 and r-Trx-BAP-Δgp160. Both lack the signal sequence (black box) and the two internal hydrophobic regions (grey boxes) of native gp160. An additional sequence consisting of thioredoxin (Trx) and biotin acceptor peptide (BAP) was fused to the N-terminus in r-Trx-BAP-Δgp160. Both proteins were provided with 6x-His tags whose positions are indicated by the asterisks. The numbers indicate the starting and ending aa residues of the native gp160 sequences retained, and fused in-frame, in the recombinant antigens. Shown to the right is design of the double antigen sandwich assay: the r-Δgp160 and r-Trx-BAP-Δgp160 antigens were labeled with Eu3+-chelate and biotin isothiocyanate, respectively. These labeling reactions (denoted by 1 and 2) produced r-Δgp160-Eu3+ (3) and r-Δgp160-Bio (4). The biotinylated capture antigen r-Δgp160-Bio was bound to SA (6) coating the microtiter well surface (5). Serum anti-HIV-1 IgG (7) and IgM (8) that are captured in the wells are revealed using the tracer, r-Δgp160-Eu3+ (3), and visualized by excitation at 340 nm followed by time-resolved measurement of fluorescence at 615 nm.

SDS-PAGE analysis of r-Δgp160 and r-Trx-BAP-Δgp160 antigens. Aliquots of total lysates of E. coli harboring the r-Δgp160 and r-Trx-BAP-Δgp160 antigen constructs, before (lanes ‘U’) and after (lanes ‘I’) IPTG induction, and aliquots of the affinity-purified antigens (lanes ‘P’), were electrophoresed on denaturing gels and visualized by Coomassie staining. Protein size markers were run in lane ‘M’; their sizes (in kDa) are shown on the left. The arrows shown on the right denote r-Trx-BAP-Δgp160 (upper) and r-Δgp160 (lower), respectively. The asterisk denotes the position of biotin ligase enzyme.
Design of double antigen sandwich TRF assays
Having obtained the pair of gp160-based recombinant antigens described above, we set up a TRF double antigen sandwich assay [20]. In this instance, r-Trx-BAP-Δgp160 was immobilized onto SA-coated plates and used to capture anti-HIV antibodies, and r-Δgp160-Eu3+ was employed in TRF assay to detect the captured antibodies. We have found in earlier work that immobilizing the biotinylated capture antigen on SA-coated plate enhances the sensitivity of the assay [29]. Surprisingly, initial assays revealed that this assay was untenable as it required very high amounts of r-Trx-BAP-Δgp160 antigen to obtain acceptable signal to background ratios. Further investigation showed this to be a result of low levels of in vivo biotinylation of this antigen (data not shown). The reason for this is not clear, though presumably it may be a reflection of the large size of the antigen, about twice as large as the in vivo biotinylated HIV-1 and −2 gp160 deletion variants reported earlier [20], and its intrinsic insolubility. It may also have been due to inaccessibility of the BAP moiety placed between the Trx and Δgp160 components of the chimeric protein. However, we think this is unlikely as similar internal positioning of BAP did not compromise biotinylation in our earlier studies [20, 21]. To compensate for the lack of in vivo biotinylation, we resorted to in vitro biotinylation of this antigen. The resultant in vitro biotinylated derivative, r-Δgp160-Bio, was used (in place of the initially intended r-Trx-BAP-Δgp160) in the double antigen sandwich TRF assay outlined above (Figure 1). Using r-Δgp160-Bio antigen immobilized on SA-coated plates, we tested sera for anti-HIV antibodies using either a one-step or two-step assay protocol. In the former, serum and r-Δgp160-Eu3+ antigen were added together into the microtiter well containing SA-immobilized r-Δgp160-Bio, whereas in the latter a wash step was introduced after serum addition, and then followed by the addition of the r-Δgp160-Eu3+ antigen. The performance of both these assays utilizing the r-Δgp160 antigen pair above, against a large panel of well-characterized sera (n=131) was analyzed as described below.
Performance evaluation of the r-Δgp160-based one- and two-step double antigen sandwich TRF assays
First, we carried out the one- and two-step assays outlined above using a panel of 59 in-house human sera that had been pre-screened with the commercially available Vidas HIV Duo Quick kit, shown in Figure 3. Based on the results with this kit, 22 sera were positive and 37 negative for anti-HIV antibodies. Remarkably, these results were mirrored exactly by both the one- and two-step r-Δgp160-based TRF assays, with no discrepancy in any of the samples whatsoever.

Analysis of an in-house sera panel using the one- and two-step double antigen sandwich TRF assays. A panel of 59 in-house assembled patient sera pre-screened for HIV-sero-status using a commercial kit (Vidas HIV Duo Quick kit, bioMérieux SA, France), were tested using the one-step (panel A) and two-step (panel B) assays. The empty and filled circles represent sera that were negative and positive, respectively, for anti-HIV-1 antibodies, as determined by the two assays.
The results with the in-house sera panel warranted additional evaluation. For this purpose, we tested several commercially procured well-characterized sera panels described earlier [20]. To begin with, the sensitivities of the one- and two-step assays were evaluated using a HIV-1 seroconversion panel consisting of 9 samples (PRB 931). Only one out of five currently used commercial kits is capable of detecting seroconversion on day 28, represented by panel member #06. The data summarized in Table 1 show that both the in-house assays picked up panel member #06 unambiguoulsy, demonstrating that they are as good as the best performing Abbott HIV-1/2 kit in this regard. The capacity to detect early seroconversion may be attributable to the inherent design of the double antigen sandwich assay format which narrows down the seroconversion window due to its ability to pick up both IgM and IgG antibodies [20, 30]. The two-step assay displayed relatively higher S/Co ratios. Next, we analyzed a worldwide panel (WWRB 302) consisting of 21 HIV-1-positive, 2 HIV-2-positive and 2 HIV-negative sera, from multiple locations in Asia, Africa, Europe, and North and South America. The data are summarized in Table 2. But for panel members #1 and #11, which were missed by the one-step assay, all other HIV-positive samples were picked out by both in-house assays. These two sera, one corresponding to HIV-1 group O (panel member #01) and the other to HIV-2 (panel member #11), were scored as weakly positive by the two-step assay. On the other hand, the HIV-2-positive panel member #25 was detectable with both the assays. Given that our antigen was not designed to pick up HIV-2, these results imply cross-reactivity of r-Δgp160 with anti-HIV-2 antibodies. Importantly, the two sera in this panel (members #10 and #30), which were HIV-negative in five different commercial assays were scored as seronegative by both our in-house assays. We then evaluated the specificity of the in-house one- and two-step assays, using a viral co-infection panel (PCA 201), consisting of 9 (of which 7 were available to us) HIV-negative and 16 HIV-positive sera samples. Many of the panel members were also seropositive for hepatitis B virus (HBV), hepatitis C virus (HCV) and/or Human T cell Leukemia virus (HTLV). Both assays recognized all HIV-1 seropositive samples correctly, irrespective of the presence of antibodies to other viruses, as shown in Table 3. It is noteworthy that of the 7 HIV-negative sera tested, 6 had evidence of other infections such as HCV, HBV or HTLV. Yet, none of these were picked up by either in-house assay, demonstrating the high degree of specificity of these assays.
The data thus far reveal that both the one- and two-step r-Δgp160-based double antigen sandwich TRF assays performed reliably when tested on three different commercially procured sera panels. Recently, we had evaluated these same sera panels in a two-step TRF assay based on a mixture of recombinant HIV-1 and HIV-2 antigens [20]. As in the present study, the earlier reported assay was also in a double antigen sandwich format. At this point, we were interested to compare the sensitivity of the earlier two-step assay with the current one- and two-step assays. To this end, we analyzed an anti-HIV-1 low titer performance panel of 15 members (PRB 108), using all three assays. The earlier assay was done as before in a two-step format, but using a single antigen, r-HIV-1env (biotinylated antigen to capture and Eu3+-chelate labeled antigen to reveal), instead of the two antigen mixture. The results of this comparative analysis are summarized in Table 4. Panel member #2 was unequivocally seronegative by the three commercial EIAs: Abbott HIV-1/2, Genetic Systems HIV-1/2 and Organon Teknika HIV-1 tests. None of the three in-house TRF assays showed reactivity with this sample. Panel member #3, a borderline sample which could not be picked up by the r-HIV-1env-based assay, was identified unequivocally by both the one- and two-step r-Δgp160-based assays. It is noteworthy that this panel member is positive for HIV-1 RNA. Another borderline sample, represented by panel member #12, was undetectable by r-HIV-1env-based assay and one-step r-Δgp160-based assay, but identified as weakly positive by the two-step r-Δgp160-based assay. Four samples (panel member #9, 10, 13 and 14) that were HIV-1 RNA positive, but scored as negative by the Organon Teknika HIV-1 EIA kit, were identified as seropositive for HIV-1 by all three in-house TRF assays. Based on the results in Table 4, the order of performance of the three assays was: r-Δgp160-based two-step assay>r-Δgp160-based one-step assay>r-HIV-1env-based two-step assay.
Collectively the data presented so far indicate that the two-step r-Δgp160-based assay performs better as an HIV-1 diagnostic test. To delineate this aspect more clearly, we compared the S/Co values obtained for the entire collection of sera analyzed in this study (n=131), using the one- and two-step r-Δgp160-based assays. This is presented in Figure 4. The mean S/Co value for the HIV positive serum samples (n = 79) using the two-step r-Δgp160-based assay was 83.1 (95% CI: 69–97.2). The corresponding value for one-step r-Δgp160-based assay was 9.7 (95% CI: 7.1-12.3). Between the two assays, the mean S/Co value was significantly higher in the two-step assay than in one-step assay (p<0.001). We interpret this as a manifestation of the high-dose Hook effect [31], wherein sera with high titers of anti-HIV antibody, in the context of the one-step assay, sequester the tracer antigen, r-Δgp160-Eu3+, thereby precluding effective double antigen sandwich formation, leading to lowered S/Co values. This anomaly is eliminated in the two-step assay by introducing an intermediate washing step which removes excess antibody after the initial capture step, before addition of the tracer antigen. However, one-step assay saves time consumed in an additional incubation step.

Comparison of the one- and two-step double antigen sandwich TRF assays. The S/Co values of all sera (n=131) analyzed in this study, using the one-step (x axis) and two-step (y axis) assay formats are included in this comparative analysis. Each circle in this plot represents one sample. The sera represented members of the in-house (red), PRB 931 (blue), WWRB 302 (green), PCA 201 (magenta) and PRB 108 (olive) panels. Filled circles represent anti-HIV positive and empty circles represent anti-HIV negative samples, as determined by the commercial tests described in the text. The dashed horizontal and vertical lines represent the cutoffs (at S/Co=1) for the two assays. Commercial panel members #1 (panel WWRB 302), #12 (panel PRB 108) and member #s 11 and 25 (panel WWRB 302), discussed in the text are indicated by ‘a’, ‘b’ and ‘c’, respectively.
Conclusions
This study has shown the feasibility of overcoming the barrier to HIV-1 gp160 expression in E. coli. It has been demonstrated that deletion of discrete hydrophobic regions can result in the expression of near full-length gp160 in E. coli hosts, in reasonably high yields. Double antigen sandwich assays in conjunction with TRF-based detection unequivocally established the diagnostic potential of the E. coli-expressed r-Δgp160 antigen. The r-Δgp160 antigen-based one- and two-step assays, were compared with several commercial HIV detection assays using a collection of 131 sera, and found to perform as well or better. Both the assays detected early seroconversion on par with the best commercial kit and in general manifested a high degree of sensitivity and specificity. The robust performance of the one-step assay suggests its potential in envisaging a rapid point-of-care assay. The two-step assay manifested a significantly higher mean S/Co value than the one-step assay, presumably by eliminating the Hook effect. The r-Δgp160 antigen provides the basis for a highly sensitive and specific HIV-1 diagnostic assay that has the potential to cut down test cost by lowering the cost of antigen production. In conjunction with luminescent lanthanide-doped inorganic nanoparticles e.g. up-converting phosphors to label the tracer antigen which can be detected with an inexpensive reader, r-Δgp160 antigen may pave the way for an affordable test in resource-poor regions where HIV-1 is prevalent.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- aa:
-
Amino acid
- BAP:
-
Biotin acceptor peptide
- CI:
-
Confidence interval
- EIA:
-
Enzyme immunoassay
- gp160:
-
Glycoprotein 160
- r-Δgp160:
-
Recombinant HIV-1 gp160 lacking hydrophobic regions as shown in Figure 1
- r-Δgp160-Eu3+ :
-
r-Δgp160 labeled with Eu3+ chelate
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV-1:
-
Human immunodeficiency virus type 1
- HTLV:
-
Human T cell leukemia virus
- IPTG:
-
Isopropyl-β-D-thiogalactopyranoside
- NAP:
-
Nucleic acid purification
- SA:
-
Streptavidin
- S/Co:
-
Signal-to-cutoff ratio
- SD:
-
Standard deviation
- Trx:
-
Thioredoxin
- r-Trx-BAP-Δgp160:
-
r-Δgp160 with N-terminal Trx-BAP fusion
- r-Δgp160-Bio:
-
in vitro (chemically) biotinylated r-Trx-BAP-Δgp160
- TRF:
-
Time resolved fluorometry.
References
Kuritzkes DR, Walker BD: HIV-1: pathogenesis, clinical manifestations, and treatment. Fields virology. Edited by: Knipe DM, Howley PM. 2007, Philadelphia: Lippincott Williams & Wilkins, 2187-2214. 5
UNAIDS world AIDS day report. 2011, (accessed on November 23, 2012) [http://www.unaids.org/en/media/unaids/contentassets/documents/unaidspublication/2011/JC2216_WorldAIDSday_report_2011_en.pdf].,
Chu C, Selwyn PA: Diagnosis and initial management of acute HIV infection. Am Fam Physician. 2010, 81 (10): 1239-1244.
Klimas N, Koneru AO, Fletcher MA: Overview of HIV. Psychosom Med. 2008, 70: 523-530. 10.1097/PSY.0b013e31817ae69f.
Stürmer M, Doerr HW, Gürtler L: Human immunodeficiency virus: 25 years of diagnostic and therapeutic strategies and their impact on hepatitis B and C virus. Med Microbiol Immunol. 2009, 198: 147-155. 10.1007/s00430-009-0117-6.
Buttò S, Suligoi B, Fanales-Belasio E, Raimondo M: Laboratory diagnostics for HIV infection. Ann Ist Super Sanita. 2010, 46 (1): 24-33.
Daskalakis D: HIV diagnostic testing: evolving technology and testing strategies. Top Antivir Med. 2011, 19 (1): 18-22.
Yang K, Li S, He F, Zhang J, Wei M, Pan H, Yang C, Xiong J, Zhang J, Xia N: Expression and purification of soluble HIV-1 envelope glycoprotein gp160 mutant from Saccharomyces cerevisiae. J Biosci Bioeng. 2009, 108 (1): 5-10. 10.1016/j.jbiosc.2009.02.009.
Sohn MJ, Lee ME, Park HS, Nham SU, Lee YI: Overexpression and purification of human immunodeficiency virus type 1 env derived epitopes in Escherichia coli. J Biotechnol. 1996, 45 (3): 211-216. 10.1016/0168-1656(95)00169-7.
Horal P, Svennerholm B, Jeansson S, Rymo L, Hall WW, Vahlne A: Continuous epitopes of the human immunodeficiency virus type 1 (HIV-1) transmembrane glycoprotein and reactivity of human sera to synthetic peptides representing various HIV-1 isolates. J Virol. 1991, 65 (5): 2718-2723.
Norrby E, Putkonen P, Böttiger B, Utter G, Biberfeld G: Comparison of linear antigenic sites in the envelope proteins of human immunodeficiency virus (HIV) type 2 and type 1. AIDS Res Hum Retrovir. 1991, 7 (3): 279-285. 10.1089/aid.1991.7.279.
Chen J, Wang L, Chen JJ, Sahu GK, Tyring S, Ramsey K, Indrikovs AJ, Petersen JR, Paar D, Cloyd MW: Detection of antibodies to human immunodeficiency virus (HIV) that recognize conformational epitopes of glycoproteins 160 and 41 often allows for early diagnosis of HIV infection. J Infect Dis. 2002, 186 (3): 321-331. 10.1086/341661.
Saville RD, Constantine NT, Cleghorn FR, Jack N, Bartholomew C, Edwards J, Gomez P, Blattner WA: Fourth-generation enzyme-linked immunosorbent assay for the simultaneous detection of human immunodeficiency virus antigen and antibody. J Clin Microbiol. 2001, 39 (7): 2518-2524. 10.1128/JCM.39.7.2518-2524.2001.
Lasky LA, Groopman JE, Fennie CW, Benz PM, Capon DJ, Dowbenko DJ, Nakamura GR, Nunes WM, Renz ME, Berman PW: Neutralization of the AIDS retrovirus by antibodies to a recombinant envelope glycoprotein. Science. 1986, 233 (4760): 209-212. 10.1126/science.3014647.
Berman PW, Riddle L, Nakamura G, Haffar OK, Nunes WM, Skehel P, Byrn R, Groopman J, Matthews T, Gregory T: Expression and immunogenicity of the extracellular domain of the human immunodeficiency virus type 1 envelope glycoprotein, gp160. J Virol. 1989, 63 (8): 3489-3498.
Nkosi SP, Huismans H, Papathanasopoulos MA: Purification and partial characterization of R5, R5X4, and X4 HIV-1 subtype C envelope glycoproteins expressed in insect cells. J Med Virol. 2005, 76 (4): 459-463. 10.1002/jmv.20383.
Fenouillet E, Miquelis R, Drillien R: Biological properties of recombinant HIV envelope synthesized in CHO glycosylation-mutant cell lines. Virology. 1996, 218 (1): 224-231. 10.1006/viro.1996.0182.
Jeffs SA, Goriup S, Kebble B, Crane D, Bolgiano B, Sattentau Q, Jones S, Holmes H: Expression and characterisation of recombinant oligomeric envelope glycoproteins derived from primary isolates of HIV-1. Vaccine. 2004, 22 (8): 1032-1046. 10.1016/j.vaccine.2003.08.042.
McDonald R, Burnett V: Novel single-round PCR and cloning of full-length envelope genes of HIV-1 may yield new insight into biomolecular antibacterial drug development. J Virol Methods. 2005, 126 (1–2): 111-118.
Talha SM, Salminen T, Swaminathan S, Soukka T, Pettersson K, Khanna N: A highly sensitive and specific time resolved fluorometric bridge assay for antibodies to HIV-1 and −2. J Virol Methods. 2011, 173 (1): 24-30. 10.1016/j.jviromet.2011.01.001.
Batra G, Talha SM, Nemani SK, Dhar N, Swaminathan S, Khanna N: Purification and characterization of in vivo biotinylated dengue virus envelope domain III based tetravalent antigen. Prot Expr Purif. 2010, 74 (1): 99-105. 10.1016/j.pep.2010.04.017.
Lode PV, Rosenberg J, Pettersson K, Takalo H: A europium chelate for quantitative point-of-care immunoassays using direct surface measurement. Anal Chem. 2003, 75: 3193-3201. 10.1021/ac0340051.
Pettersson K, Lode PV, Eriksson S, Lövgren J, Takalo H: Multi-assay point of care platform: highly sensitive time-resolved fluorometric detection in combination with a universal “All-in-one” assay format. Point of care. 2003, 2: 225-232.
Mukkala V-M, Hänninen E, Hemmilä I: Synthesis of two biotin isothiocyanate derivatives. Proceedings of 8th European symposium on organic chemistry. 1993, Barcelona, Spain: Book of abstracts. Universitat Autónoma de Barcelona, Sitges, 86-
Pancera M, Majeed S, Ban YEA, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD: Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA. 2010, 107 (3): 1166-1171. 10.1073/pnas.0911004107.
Finzi A, Pacheco B, Zeng X, Kwon YD, Kwong PD, Sodroski J: Conformational characterization of aberrant disulfide-linked HIV-1 gp120 dimers secreted from overexpressing cells. J Virol Methods. 2010, 168 (1–2): 155-161.
Lin CH, Lin CH, Chang CC, Wei TS, Cheng SF, Chen SS, Chang DK: An efficient production and characterization of HIV-1 gp41 ectodomain with fusion peptide in Escherichia coli system. J Biotechnol. 2011, 153 (1–2): 48-55.
Vogel EP, Curtis-Fisk J, Young KM, Weliky DP: Solid-state nuclear magnetic resonance (NMR) spectroscopy of human immunodeficiency virus gp41 protein that includes the fusion peptide: NMR detection of recombinant Fgp41 in inclusion bodies in whole bacterial cells and structural characterization of purified and membrane-associated Fgp41. Biochemistry. 2011, 50 (46): 10013-10026. 10.1021/bi201292e.
Batra G, Nemani SK, Tyagi P, Swaminathan S, Khanna N: Evaluation of envelope domain III-based single Chimeric tetravalent antigen and monovalent antigen mixtures for the detection of anti-dengue antibodies in human sera. BMC Infect Dis. 2011, 11: 64-10.1186/1471-2334-11-64.
Siitari H, Turunen P, Schrimsher J, Nunn M: New sensitive and specific assay for human immunodeficiency virus antibodies using labeled recombinant fusion protein and time-resolved fluoroimmunoassay. J Clin Microbiol. 1990, 28 (9): 2022-2029.
Butch AW: Dilution protocols for detection of hook effects/prozone phenomenon. Clin Chem. 2000, 46 (10): 1719-1721.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/12/325/prepub
Acknowledgements
The authors thank Dr. Tytti Vuorinen from Department of Virology, University of Turku, Turku, Finland, for providing human serum samples. This work has been carried out under a bilateral Indo-Finnish collaborative research programme, and was supported by grants from the Finnish Funding Agency for Technology and Innovation (TEKES), Finland, and Department of Biotechnology, Government of India.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SMT, SKN, TSa and SK conducted the experiments. SMT, SS, TSo, KP and NK designed the study and wrote the initial draft. SMT and SS wrote the final manuscript. All the authors read and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Talha, S.M., Nemani, S.K., Salminen, T. et al. Escherichia coli–expressed near full length HIV-1 envelope glycoprotein is a highly sensitive and specific diagnostic antigen. BMC Infect Dis 12, 325 (2012). https://doi.org/10.1186/1471-2334-12-325
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-12-325