
Abstract
Background
The thalassemic syndromes originate from mutations of the globin genes that cause, besides the characteristic clinical picture, also an increased Hb F amount. It is not yet clear if there are more factors, besides the beta globin genotype, determining the Hb F production. We have tried to find out if there are relations between total Hb and Hb F, between erythropoietin (Epo) and Hb F, between Hb F and point mutations of the gamma gene promoters.
Materials and Methods
Hematologic parameters, iron status, alpha/non-alpha globin ratio, Epo level, and thalassemic defects of the alpha-, beta-, and gamma-globin genes were explored using standard methods in patients affected by thalassemic diseases. Ninety-five non thalassemic individuals have been examined as controls.
Results
Two clinical variants of beta-thalassemia intermedia referred to as beta-thal int sub-silent and evident are associated with distinct sets of mutations of the beta-globin gene. Silent beta thal mutations are invariably associated with sub-silent beta thal int; beta° or severe beta+ thal mutations are associated with evident beta thal int (88%) and almost invariably (98%) with thalassemia major. A positive correlation was observed between the severity of the disease and the Hb F level, but no correlation was found between the Hb F and erythropoietin (Epo) level. The mutation Ggamma -158 C→T was detected in 26.9% of patients affected by beta-thal int sub-silent and evident, respectively, but only in 2% of patients with thalassemia major.
Conclusions
The severity of beta-thal int and the increased Hb F level are strictly dependent from the type of beta-globin gene mutations. No relation is found between Hb F synthesis and Epo secretion. The mutation Ggamma -158 C→T, common among patients affected by beta-thal int and very rare in thal major patients, does not seem, in this study, to influence the Hb F content in beta thal int patients.
Similar content being viewed by others


Background
The beta-thalassemia syndromes (thalassemia major and beta-thalassemia intermedia) originate from the absence of or from a reduction in the synthesis of structurally normal beta-globin chains. The beta-globin chain deficiency contributes to the pathogenesis of anemia by impairing the formation of functional Hb A and by causing ineffective erythropoiesis. The anemia stimulates the secretion of Epo and leads to erythroid hyperplasia of the bone marrow and to an accelerated erythropoiesis [1]. As a result, erythroblasts expressing the fetal globin gene are generated and fetal hemoglobin (Hb F) is synthesized in the postnatal life [2]. Two more factors, the amplification of the cellular clones producing F-cells and the slower turnover of these cells in circulating blood favour the Hb F presence in thalassemia [3]. In these affections the persistent synthesis of Hb F is very important because it restores the alpha/non-alpha globin ratio, thus reducing the bone marrow hyperplasia and ineffective erythropoiesis and therefore the severity of disease.
Genetically, the beta-thalassemia syndromes are heterogeneous. The severity of these disorders is determined primarily by the type of the gene defect as well as the gene dosage.
The molecular analysis has uncovered that different clinical variants of beta-thalassemia are associated with a wide spectrum of different mutations of the beta-globin gene; thalassemia major patients, characterized by a severe transfusion-dependent anemia and a marked increase in the synthesis of Hb F, are either homozygous carriers of mutations that abolish the beta-globin synthesis (beta° thai) or compound heterozygotes for beta° thal and severe beta+ thal defects which drastically reduce it. These mutations are referred to as severe beta-thal mutations. beta-thal int patients which have a less severe anemia and a modest increase in the synthesis of Hb F, are carriers mainly of beta+ thal mutations that reduce slightly beta-globin synthesis. These mutations are referred to as mild beta-thal mutations.
Furthermore, deletions of the non-alpha globin gene cluster encompassing the beta- and delta-globin genes are associated with a genetic variant of beta-thalassemia (deltabeta thal), which is characterized by a marked increase in the synthesis of Hb F even in the heterozygous state. In these patients the gamma-globin genes remain intact and are expressed in the postnatal life, thus reducing the ineffective erythropoiesis and ameliorating the severity of the disease.
It is worth noting that also genetic factors unlinked to the beta globin cluster, favour the Hb F synthesis in the post-fetal life [4]. Particularly interesting for its frequency is the polimorphic single-base substitution C→T at Ggamma -158, recognizable for the creation of a XmnI restriction site, that appears to cause an increase of Hb F expecially during erythropoietic stress.
Recently, it has been suggested that the increased synthesis of Hb F associated with beta-thalassemia syndromes is directly or indirectly dependent from the Epo level [5, 6].
The aim of this study was to investigate the relationships between the genetic defects and the severity of the disease in patients affected by a thalassemic disease. In particular, the relationships between the genetic defect, the total Hb, and between the synthesis of Hb F and the Epo level have been examined in patients with beta thalassemia intermedia (beta thal int). In addition, the distribution and frequency of point mutations of the promoter region of gamma-globin genes were analysed and the relationship between these mutations and the content of Hb F was investigated. The analysis of the beta-globin gene and gamma-globin gene mutations was extended also to patients affected by Cooley's disease.
Subjects
Seventy-four patients with clinical picture of beta thal int have been examined: 33 of them were affected by a mild or sub-silent form of the disease, and 41 by an evident clinical picture (Tab. 1). In addition, 58 patients carried a beta thal defect and also a triplicated or quadruplicated alpha locus. Twenty-four patients were affected by thalassemia major, and 15 by Hb H disease. The control group included 53 healthy individuals and 42 iron-deficient, non thalassemic patients.
Patients with an homozygotic condition for a delta beta thal deletion were excluded from this study, since only Hb F is synthesized. The patients homozygotes for severe beta-globin defects, and carriers of an alpha thal defect, were also excluded.
All of the individuals examined in this study were in the post-puberal age.
Methods
Blood samples were collected in EDTA. Hematologic parameters were determined using a Technicon H1 Analyser (Bayer). Hb A2, Hb F and Hb H values were determined using an electrophoretic method [7]. Concentrations of Hb F higher than normal were confirmed by radial immunodiffusion (Quick Plate kit, Helena Laboratories, Beaumont, TX, USA). Serum iron and ferritin levels were measured by standard methods. The serum level of erythropoietin (Epo) was determined using the Epo-Trac kit (INSTAR Corporation, Stillwater, MN, USA). The average value of 53 healthy individuals was assumed as the normal Epo concentration. Reticulocyte counts were performed on fresh blood preparations stained with brilliant-cresyl-blue. The synthesis of globin chains in vitro was determined following the method of Clegg [8], using a high pressure liquid chromatograph.
The Amplification Refractory Mutation System (ARMS) was used to detect mutations of the beta- and gamma-globin genes [9, 10] and a modified PCR method previously described in our laboratories [11], was used to detect alpha-globin genes defects. The patients affected by thalassemia major or intermedia were screened for all of the beta- and alpha-globin gene mutations present in the Italian population (Tab. 4, ref. [4, 12]) and the mutations analysis was also extended to mutations of the promoter region of the gamma-globin genes which are until reported to occur in the Italian population (Ggamma-158 C→T; Agamma -196 C→T; Ggamma -175 T→C; Agamma -117 G→A; AgammaT and the deletion Agamma -225/-222 bp). The Ggamma -158 C→T and AgammaT substitutions were searched in all of the subjects reported in Table 1. The remaining mutations of the gamma genes promoters were searched in 286 more chromosomes of beta-thalassemia patients or control subjects.
The statistical significance was evaluated using the Student's t-test. Linear regressions were fitted by the least square deviations method and the correlation coefficient r was calculated according to a standard formula. A p value < 0.05 was assumed to be statistically significant, as only few tests were performed.
Results
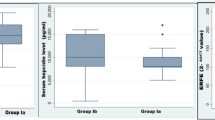
The analysis of the hematologic data and the clinical symptoms of 74 patients affected by beta-thal int indicate that this disease can be classified into two clinical variants referred to as sub-silent and evident beta-thal int (Table 1). The patients with sub-silent beta-thal int have a reduced total Hb content and show no or very mild clinical symptoms. The patients with evident beta-thal int show severe clinical symptoms, often associated with massive splenomegaly, and 41% of them have undergone splenectomy. The Hb content is significantly lower than that of the patient with sub-silent beta-thal int (t = 7.44; gl 72; p < 0.001). The Hb F level is higher in with evident than in patients with sub-silent beta-thal int, and the difference is statistically significant (t = 2.28; gl 72; p < 0.05). Thus, the severity of clinical symptoms of beta-thal int correlates with a decrease of the total Hb content and an increase of the Hb F content. Normally the Hb F content is positively correlated with the total Hb content, as illustrated in Fig 2.

Epo level and Hb F content are not related in patients with β-thalassemia intermedia. The logarithm of the Epo level was plotted against the corresponding total Hb content of each patient. The patients showing an increase of Hb F higher than 40% are represented by red squares, whereas the blue squares denote the patients showing an increase of Hb F equal to or less than 40%.
In the 42 iron-deficient but not thalassemic subjects, the Epo level ranging from 2.7 to 1960 mU/1 with a mean value of 160.10 ± 312.60 mU/1, was negatively correlated (r = -0.74) to the Hb content (Fig. 1). The correlation is statistically significant (n = 42; p < 0.001).

Negative correlation between total Hb content and Epo level in iron-deficient patients. The logarithm of the Epo level plotted against the corresponding total Hb content of each patient is denoted by a blue square. The linear regression (solid line) fitting the data is described by the following equation: log [Epo] = 4.78–0.33 [Hb]. The correlation coefficient r is -0.74.
The mean Epo level of beta-thal int patients is apparently lower than that of iron-deficient patients (Tab. 1). Regarding to total Hb, only between the groups of sub-silent beta-thal int patients and iron-deficient patients the difference is statistically significant (t = 2.17; gl 73; p < 0.05).
The mean values of the Epo level and the Hb F content in the two groups of beta-thal int seem to be positively related, but an accurate analysis of single cases indicates (Tab. 2) that the Epo level and Hb F content are unrelated: subjects showing a low content of Hb F and an high level of Epo as well as subjects showing an high content of Hb F and a low level of Epo were frequently observed. This observation is reinforced by the analysis of groups of subjects in which the severity of anemia is similar. Within the group of evident beta tal int patients (the only adapt for this analysis) subjects showing an Hb F level higher or lower than 40% [5, 6] were selected. As illustrated in Fig. 2 and Tab. 3, subjects having a very similar Epo level and Hb content fall in both classes of Hb F content, indicating that Hb F content and Epo level are not related.
Different sets of mutations of the globin genes are associated with groups of patients characterized by a different severity of the disorder. The spectrum of the beta-globin gene mutations commonly found in the Italian population can be divided in three categories, the first of which includes the -101 C→T, -92 C→T, and IVS II 844 C→G single nucleotide substitutions; the second the -87 C→G, -86 C→G or C→A, and IVS I C→T single nucleotide substitutions, and the third the beta° 39 C →T, IVS I 110 G→A, IVS II 745 C→G single nucleotide substitutions and the frameshift 6 (-A) mutation. These three categories are referred to as silent beta-thal, mild beta-thal, and severe beta-thal mutations, respectively.
In the group of 33 patients with sub-silent beta-thal int the frequencies of silent beta-thal, mild beta-thal, and severe beta-thal mutations were 37%, 13% and 50%, respectively (Tab. 4). In the group of 42 patients with evident beta-thal int, the most common and equally frequent (44%) beta-globin gene defects were mild and severe beta-thal mutations. The frequency of silent beta-thal mutations was 12%. Conversely, severe beta-thal mutations were found at high frequency (83%) in thalassemia major patients. Silent beta-thal mutations were detected only in a small fraction (2.1%) of thalassemia major patients.
In the 58 patients both heterozygous for a mutation of the beta-globin gene and heterozygous or rarely homozygous for the presence of three or four copies of one alpha-globin locus, the beta-globin gene defects were invariably beta° or severe beta+ thal mutations (Tab. 4 and ref. [13]); the alpha-globin gene mutation was in 54 out of 58 patients a triplicated alpha anti 3.7 I gene cluster; in 3 patients a quadruplicated alpha anti 4.2 gene cluster, and in one patient the homozygous condition for a triplicated alpha anti 3.7 I gene cluster. In these patients, the average Hb content was 10.0 ± 1.38 g/dl (Table 1), the content of Hb F was slightly higher than normal, ranging from 2.0 to 19.3 with an mean value of 5.15 ± 3.9 %, and the average Epo level was 28.45 mU/1, ranging from 7.85 to 60.72 mU/l. This value is lower than that of regularly transfused thalassemia major patients. Although the Hb F content appears to be low in all these patients, their clinical picture is highly heterogeneous, 50% of the patients having a sub-silent beta thal int, 30% an evident beta thal int and 20% a severe anemia with 5 transfusion dependent patients. Ten out of 58 patients (17.2%) have undergone splenectomy.
The patients with Hb H disease have a normal Hb F level, and a Epo level lower than that of patients with beta-thal int.
In the beta-thalassemia patients and in the control subjects, screened for the six mutations of the promoter region of the gamma-globin gene observed in Italy, only the Ggamma -158 C→T and AgammaT single nucleotide substitutions were detected. In all cases except 7 the AgammaT mutation was to the Agamma -225–222 bp deletion. The frequencies of the Ggamma -158 C→T and AgammaT mutations are significantly different in the various groups of beta-thalassemia patients (Tab. 5), with the lowest frequency of the Ggamma -158 C→T mutation (2.1%) in thalassemia major patients.
In the group of patients with evident beta-thal int the frequency of the Ggamma -158 C→T mutation was 6.2% in 7 patients having a total Hb content > 9 g/dl and 1.6% in 32 patients having a total Hb content ≤ 9 g/dl.
Discussion
It is well known that the severity of thalassemic diseases (thalassemia major and thal int) syndromes is primarily determined by the nature and type of mutations as well as gene dosage of the globin genes underlying these disorders. The results reported in this study extend this conclusion to two clinical variants of beta-thal int, referred to as sub-silent and evident beta-thal int. Distinct sets of mutations of the beta-globin gene have been found associated with these two variants of beta-thal int. Silent mutations of the beta-globin gene were detected at high frequency in patients affected by sub-silent beta-thal int, whereas mild and severe mutations of the beta-globin gene were the most common mutations amongst the patients affected by evident beta-thal int (Table 4). This notion is reinforced by the high frequency (83%) of severe beta+ and beta° mutations in patients affected by thalassemia major.
Several hematologic parameters of beta-thalassemia reflect the diversity of mutations at beta-globin locus. The total Hb content was found to be significantly different between patients affected by thalassemia major and patients with beta thal int, as well as between patients affected by sub-silent and patients with evident beta-thal int (Table 1). These last two groups of patients showed also significant differences in the content of Hb F (t = 2.28; df = 72; p < 0.05).
The comparative analysis of the serum Epo level in groups of patients affected by thalassemia diseases or iron deficiency shows first of all that the synthesis of Epo, as predicted, is always directly related to the severity of anemia and, consequently, inversely related to the total Hb content. Furthermore, in the thalassemic diseases mean Epo level is consistently lower than that of iron deficient patients, probably as a result of the different pathophysiology of the anemia in the two conditions. No firm relationship could be established between the Epo and Hb F levels. The variations of these two parameters in each patient with evident beta thal int appear to be erratic (Tab. 2); and in patients having a similar level of both total Hb and Epo, the amount of Hb F is indifferently higher or lower than 40% (Tab. 3). Therefore, the data do not support the hypothesis that Hb F content is regulated by the serum level ofEpo. The relationship between total Hb content, Hb F content and Epo level were not studied in patients with Hb H disease, since Hb F is in these patients below the detection limit.
In patients with a compound heterozygosity for a triplicated copy of a alpha globin gene cluster and a beta globin gene mutation, only a moderate increase of the Hb F level (mean 5.5 ± 3.9% within a range from 2.0 to 19.3%) was observed. This observation indicates that the excess of alpha globin chains, as assessed by the unbalanced alpha/non-alpha globin ratio (2.5) is not sufficient to increase the gamma-globin chains synthesis.
Only the Ggamma -158 C→T and AgammaT single nucleotide substitutions out of 6 gamma gene promoter region mutations explored in this study were detected, and their frequencies varied in different groups of patients (Table 5). The role of this mutation in determining the Hb F amount is not yet definitively cleared. The demonstration, already reported by other Authors [14], that the frequency of Ggamma -158 mutation is very low in beta thal major patients (Tab. 5), and, inside the beta thal int patients, higher in those with a higher amount of total Hb and lower in those with a lower amount of total Hb, seems to indicate that this mutation is important in ameliorating the severity of the disease.
Conversely, other obstervations [15–17] report that this mutation does not influence the Hb F content of subjects which are heterozygous for a beta thal mutation [16] as well as of heterozygous compounds for a beta thal mutation and a triplicated or quadruplicated copy of the alpha-globin cluster.
At variance with these observations, in sickle cell disease patients carrying some betas haplotypes which show a high frequency (up to 100%) of the Ggamma -158 C→T mutation, show also an increase of the Hb F content that leads to a reduction of the severity of the disease [18].
The relationship between the content of Hb F and point mutations of the promoter region of the gamma-globin genes in thalassemic patients needs then to be further investigated. The analysis must be extended to those mutations of the gamma-globin gene promoter region that have not yet been detected in the population of Italy or other Mediterranean countries. The hypothesis [19] that the Ggamma -158 C→T mutation when linked in cis to the sequence motif (AT)x(T)y mapping at position -530 in the 5' untranslated region of the beta-globin gene could interact to generate a high expression of Hb F under the erythropoietic stress, has to be ulteriorly evaluated. Finally, the role played by loci unlinked to the beta-globin locus in the regulation of Hb F synthesis remains to be elucidated. Two candidate regulatory loci have been mapped at Xp22 [FCP locus [20, 21]] and 6q22.3-23.2 [22].
A study regarding all these factors, obviously necessary also for the various heterozygotic thalassemic conditions, is now in progress in our laboratories.
Conclusions
The severity of beta-thalassemia syndromes including the increased synthesis of Hb F is primarily determined by the genotype of the beta-globin locus. The synthesis of Hb F does not appear to be regulated by the serum level of Epo. The Ggamma -158 C→T mutation is frequent among patients with beta-thal int, and very rare in patients with thalassemia major, but at variance with sickle cell disease, increased synthesis of Hb F in beta-thal int patients cannot be firmly related to the linkage in cis of this mutations.
References
Nienhuis AW, Barker JE, Croissant RD: Overview: mechanims of the regulation of hemoglobin synthesis at the cellular level. Ann. N.Y. Acad. Sci. 1980, 344: 189-205.
Silvestroni E, Bianco I, Modiano G, Vullo C: Talassemie. Trattato Italiano di Medicina Interna, diretto da P. Introzzi, II Edizione, USES Ed. Scient, Firenze. 1982
Weatherall DJ, Clegg JB: The Thalassaemia Syndromes. 1981, Blackwell Scientific Publication, Third
Bianco Silvestroni I: Le talassemie. Un problema medico-sociale: ieri e oggi. Collana Studi e Ricerche. 1st. It. Med. Soc. Ed. 1998
Galanello R, Barella S, Ideo A, Gasperini D, Rosatelli C, Paderi L, Paglietti E, Sollaino C, Perseu L, Loi D, Cao A: Genotype of Subjects With Borderline Hemoglobin A2 Levels: Implication for beta-Thalassemia Carrier Screening. Am. J. Hematol. 1994, 46: 79-81.
Camaschella C, Gonella S, Calabrese R, Vischia F, Roetto A, Graziadei G, Mazza U, Cappellini MD: Serum erythropoietin and circulating transferrin receptor in thalassemia intermedia patients with heterogeneous genotypes. Haematologica. 1996, 81: 397-403.
Graziani B, Bianco I, Carboni C, Lerone M, Valente M, Sidorini B, Ricotti G, Bianco P: Tecniche e strategie di screenings di massa delle microcitemie. VI Congr Intern su "La Prevenzione delle Malattie Microcitemiche". Roma Ed. Min. Med., Pag. 1980, 185-9.
Clegg JB: Hemoglobin synthesis. Methods in Hematology: The Thalassemias. Churchill Livingstone, London. Edited by: DJ Weatherall. 1983
Old JM, Varawalla NY, Weatherall DJ: Rapid detection and prenatal diagnosis of beta-thalassaemia: studies in Indian and Cypriot population in the UK. Lancet. 1984, ii: 834-
Bianco I, Cappabianca MP, Foglietta E, Morlupi L, Deidda G, Graziani B: Difetti molecolari nelle beta microcitemie e correlazioni genotipo-fenotipo. Progr. Med. 1995, 51: 1-22.
Foglietta E, Deidda G, Graziani B, Modiano G, Bianco I: Detection of alpha-globin gene disorders by a simple PCR methodology. Haematologica. 1996, 81: 387-396.
Cappabianca MP, Morlupi L, Rinaldi S, Graziani B, Bianco I: Le beta microcitemie nel Lazio: varietà molecolari ed incidenze. Progr. Med. 1995, 51: 41-
Bianco I, Lerone M, Foglietta E, Deidda G, Cappabianca MP, Morlupi L, Ponzini D, Grisanti P, Di Biagio P, Amato A, Mezzabotta M, Graziani B: Phenotypes of individuals with a beta thal classical allele associated either with a beta thal silent allele or with alpha globin gene triplication. Haematologica. 1997, 82: 513-525.
Thein SL, Wainscoat JS, Sampietro M, Old JM, Cappellini D, Fiorelli G, Modell B, Weatherall DJ: Association of thalassaemia intermedia with a beta-globin gene haplotype. Br. J. Haematol. 1987, 65: 367-73.
Kanavakis E, Metaxotou-Mavrommati A, Kattamis C, Wainscoat JS, Wood WG: The triplicated alpha gene locus and beta thalassaemia. Br. J. Haematol. 1983, 54: 201-
Traeger-Synodinos J, Kanavakis E, Vrettou C, Maragoudaki E, Michael T, Metaxotou-Mavrommati A, Kattamis C: The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: clinical, haematological, biosynthetic and molecular studies. Br. J. Haematol. 1996, 95: 467-10.1046/j.1365-2141.1996.d01-1939.x.
Vrettou C, Kanavakis E, Traeger-Synodinos J, Metaxotou-Mavrommati A, Basiakos I, Maragoudaki E, Stamoulakatou A, Papassotirou I, Kattamis C: Molecular Studies of beta-Thalassemia Heterozygotes With Raised Hb F Levels. Hemoglobin. 2000, 24: 203-
Nagel RL, Ranney HM: Genetic Epidemiology of Structural Mutations of the Beta-Globin Gene. Seminars in Hematol. 1990, 27: 342-
Ragusa A, Lombardo M, Beldjord C, Ruberto C, Lombardo T, Elion J, Nagel RL, Krishnamoorthy R: Genetic Epidemiology of Beta-Thalassemia in Sicily: Do Sequences 5' to the Ggamma Gene and 5' to the Beta Gene Interact to Enhance HbF Expression in Beta-Thalassemia?. Am. J. Hematol. 1992, 40: 199-206.
Dover GJ, Smith KD, Chang YC, Purvis S, Mays A, Meyers DA, Sheils C, Serjeant G: Fetal Hemoglobin Levels in Sickle Cell Disease and Normal Individuals Are Partially Controlled by an X-Linked Gene Located at Xp22.2. Blood. 1992, 80: 816-24.
Miyoshi K, Kaneto Y, Kawai H, Ohchi H, Niki S, Hasegawa K, Shirakami A, Yamano T: X-Linked Dominant Control of F-Cells in Normal Adult Life: Characterization of the Swiss Type as Hereditary Persistence of Fetal Hemoglobin Regulated Dominantly by Gene (s) on X Chromosome. Blood. 1988, 72: 1854-
Craig JE, Rochette J, Sampietro M, Wilkie AOM, Barnetson R, Hatton CSR, Demenais F, Thein SL: Genetic Heterogeneity in Heterocellular Hereditary Persistence of Fetal Hemoglobin. Blood. 1997, 90: 428-
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2326/2/2/prepub
Acknowledgements
The Authors are grateful to Dr. Giacomo D'Agostaro (ENEA, Rome) for his cooperation and helpful comments in preparing the manuscript; Dr. Paolo Cianciulli Director of the Day Hospital for Thalassemics of the Sant'Eugenio Hospital of Rome, to have made possible the study of his thalassemic patients; to the Mrs. Brunella Sidorini and Mrs. Nadia D'Arcangeli for their technical assistance; to Mr. Marco Ballante for his excellent contribution to the making of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Mastropietro, F., Modiano, G., Cappabianca, M.P. et al. Factors regulating Hb F synthesis in thalassemic diseases. BMC Hematol 2, 2 (2002). https://doi.org/10.1186/1471-2326-2-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2326-2-2