
Abstract
Background
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to be effective in inhibiting colorectal cancer. Cyclooxygenase activity is thought to mediate, in part, this cancer preventive effect. From observations made when cells that express cyclooxygenase activity were treated with NSAIDs and known cancer preventive agents, I have postulated that arachidonic acid (AA) release is associated with cancer prevention. In this study, the effects of NSAIDs on two cells that do not express cycloxygenase activity are detailed.
Results
NSAIDs and several cancer preventive agents release AA from human colon cancer cells (the HCT-15 cell line). The concentrations of NSAIDs required to release significant amounts of AA from the HCT-15 cells are greater than those required to inhibit the lactacystin plus 12-0-tetradecanoyl-13-acetate stimulated cyclooxygenase activity of rat liver cells. NSAIDs, tamoxifen and simvastatin were found to hemolyze erythrocyte cells which also do not express cyclooxygenase activity
Conclusion
The data demonstrate that AA release is independent of cyclooxygenase activity and together with hemolysis suggest that intercalation of the plasma membrane by some NSAIDs and cancer preventive agents, e.g. tamoxifen, mediates this release. A mechanism by which many of these drugs affect several diverse biologic properties including deesterification of membrane phospholipids by phospholipases to release AA is presented.
Similar content being viewed by others

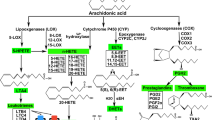
Background
Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase (COX) activity and the local production of COX products that affect inflammation [1]. Nevertheless, the role of NSAIDs in several biologic pathways, at high doses, has been questioned [2, 3]. Some NSAIDs have been shown to be cancer preventive [4, 5]. The role of COX in cancer prevention is suggested by the findings that COX is overexpressed in tumors and that PGE2 levels are increased in tumors. COX independent mechanisms for cancer prevention have been shown by Rigas and collaborators [6, 7].
Based on the stimulation of AA release by several known cancer preventive agents, e.g., tamoxifen, 9-cis-retinoic acid, vitamin D3, statins, anti-oxidants found in green tea and red wine, and peroxisome proliferator-activated receptor ligands, I have proposed that AA release by cells is associated with cancer prevention [8–10]. Celecoxib, a COX inhibitor, releases AA from rat liver, human colon carcinoma, and rat glial cells in culture. Three of these cells, rat liver (the C-9 cell line), the human colon cancer (the HT-29 cell line) and rat glia (the C-6 cell line) express COX [6, 11, 12]. The human colon carcinoma cell line (HCT-15) does not produce prostaglandins (PG) when treated with know stimulators of PG synthesis such as mellitin, A-23187, fetal bovine serum or exogenous AA. Nor do these cells express COX as measured by Northern Blotting (6). In this report, evidence is presented that NSAIDs and other cancer preventive agents release AA from these HCT-15 cells.
Primary cultures of gastric mucosal cells undergo necrosis as well as apoptosis after incubation with NSAIDs [13]. Thus, the effects of NSAIDs on the morphology of sheep erythrocytes (SRBC), cells that also do not express COX [14, 15] were also examined using lysis as an indicator of membrane perturbation and necrosis. Some NSAIDs as well as tamoxifen and simvastatin hemolyze the washed COX negative SRBC. Several of the NSAIDs are known to intercalate the plasma membrane of cells [2, 3].
Results
Preliminary experiments showed that the release of AA from HCT-15 cells by 50 μM celecoxib was complete after a 19 h incubation. The release of AA from these cells by the COX inhibitors, celecoxib (50 μM) and NS398 (52 μM), is shown in Table 1. Celecoxib is more effective than NS398 at comparable doses. The concentration dependencies of the release by celecoxib, sulindac sulfide, indomethacin and ibuprofen are shown in Fig. 1. Celecoxib is 40 to 100 times more effective than ibuprofen, and more effective than sulindac sulfide or indomethacin. In contrast to the observations presented in this report, indomethacin, 200 μM, inhibited unstimulated AA release from rabbit kidney slices 50% [16]. At 15 μM, indomethacin did not inhibit AA release from perfused rabbit kidney after a 1 h incubation, but did inhibit its rate of release after incubation with angiotensin II [16]. In our studies, indomethacin stimulates AA release from unstimulated human colon carcinoma (HCT-29 cells), rat liver (C-9 cells), rat glial (C-6 cells) [8, 9, 17, 18] and human colon carcinoma (HCT-15 cells).

Release of AA from HCT-15 cells by increasing quantities of celecoxib (O), sulindac sulfide (◇), indomethacin (□) or ibuprofen (△). The HCT-15 cells were incubated with the NSAIDs for 19 h. Centrifuged culture fluids (200 μl) were counted for radioactivity.
The dose-dependencies of AA release by tamoxifen and simvastatin as well as those of thapsigargin and tetrandrine are shown in Fig. 2. Thapsigargin is active even at 1.0 μM. It stimulates AA release from HT-29 cells [18] and induces apoptosis in androgen-independent prostate cancer cell line [19]. Its analogs exhibit chemoprevention of androgen-independent prostate cancer [20]. Thapsigargin also induces apoptosis of human neuroblastoma, colon cancer and thymocytes [21, 22]. Tetrandrine stimulates AA release from HT-29 cells [18] and is known to induce apoptosis and is chemopreventive [23]. The dose-response curve of tetrandrine is markedly more shallow than those of the NSAIDs examined and suggests a different mechanism of AA release.

Release of AA from HCT-15 cells by various concentrations of thapsigargin (□), tamoxifen (△), simvastatin (○) and tetrandrine (◇). The experiment was done with triplicate dishes. Incubation conditions were similar to those described in Fig. 1.
The insolubility of several of these drugs precludes generation of extensive dose-response curves at high concentrations. At concentrations at which they were soluble, they release a significant amount of AA (Table 2). The concentrations of NSAIDs required for release of AA are greater than those required to inhibit COX as measured by PGI2 production of lactacystin plus TPA stimulated COX positive rat liver cells (data not shown). Indomethacin, 50 μM, significantly stimulates AA release from the HCT-15 cells, but 0.003 μM, 4 orders of magnitude less, is required to inhibit 50 % of PGI2 produced by different cells (the COX positive C-9 rat liver cells) stimulated by lactacystin plus TPA treatment. COX-inhibition depends on several factors including time of incubation, cellular source of the COX, stimulator of the COX (endotoxin, A-23187, TPA, etc.), subcellular location of the COX, concentration of substrate and the degree of COX-PLA2 coupling. For example, the IC50 for COX-2 inhibition of PG production by indomethacin when treated with endotoxin-stimulated broken macrophage was 1.1 μM, but when assayed with purified enzyme, it was 14 μM [24].
A low concentration of SRBC (6.5 × 107 cells/ml) was used to quantitate hemolysis [25] after incubation of these COX negative cells with several NSAIDs, tamoxifen or simvastatin. Hemolysis by celecoxib (80 μM) and tamoxifen (20 μM and 40 μM) is shown in Table 3. The concentration dependencies for hemolysis by celecoxib, sulindac sulfide, indomethacin and ibuprofen are shown in Fig. 3. As with AA release (Fig. 1), celecoxib is the most effective. Treatment of cells with tamoxifen and simvastatin also hemolyze the SRBC (Fig. 4). Tetrandrine or thapsigargin, even when tested at 1 mM and 100 μM respectively do not lyse the SRBC (data not shown), suggesting that the mechanisms of AA release by tetrandrine and thapsigargin differs from that of NSAIDs, tamoxifen or simvastatin.

Hemolysis of 6.5 × 107 SRBC (2.6 × 107/ml) by various concentrations of celecoxib (ɹ), sulindac sulfide (◇), indomethacin (○) and ibuprofen (△). Experimental conditions were similar to those described in Table 4.

Hemolysis of 6.5 × 107 SRBC (2.6 × 107/ml) by various concentrations of tamoxifen (□) and simvastatin (◇). Experimental and controls are described in Table 4.
When the effect of cell density on hemolysis was tested with celecoxib, tamoxifen or simvastatin, the number of cells lysed was higher at the lower cell densities (data not shown). Under the experimental conditions described, an approximate number of molecules of drug required to hemolyze 2.5 × 107 SRBC was calculated (Table 4). These values may be grossly overestimated since binding to the 1 % bovine serum albumin (BSA) in the vehicle was not considered.
Discussion
In contrast to HT-29 cells which are known to be COX dependent, HCT-15 cells do not respond to known stimulants of COX activity, such as mellitin, A-23187, fetal bovine serum or even exogenous AA [6]. Nor are transcripts of mRNA from both COX-1 and COX-2 genes detected in these HCT-15 cells [6]. When measured by confocal microscopy, however, COX is detected in these HCT-15 cells [26]. A study of AA release by the COX-null embryo fibroblasts described by Zhang et al [27] would be informative and provide another COX negative cell for study. Although erythrocytes, are COX negative [14, 15], they may express lipoxygenase (LOX). Sulindac sulfide and indomethacin stimulate AA release from the colorectal cancer cell lines, SW480 and HCT-116. It is the AA release per se that leads to apoptosis [28]. Chan et al [28] have suggested that inhibition of COX is a mechanism of action of NSAIDs. By inhibiting COX, more AA is made available as substrate for sphingomyelin to ceramide conversion. I have proposed that the release of AA is associated with cancer prevention [8–10, 17, 18]. The AA release was observed from COX positive rat liver (C-9), COX positive human colon cancer (HT-29) and COX positive rat glioma (C-6) cells. Growth of COX negative and COX positive cells is inhibited and their progression to apoptosis is increased by NSAIDs [6, 7]. In cells different than those cited above, others have demonstrated COX independence for NSAID actions [reviewed in [29, 30]].
Red blood cells do not express COX activity [14, 15], yet incorporate AA into their phospholipids. After a 5 min incubation with the Ca2+ ionophore, A-23187, 26 % the AA is found unesterified when analyzed by high pressure liquid and thin layer chromatography [14]. In the present study, hemolysis is observed after a 19 h incubation of SRBC with NSAIDs, tamoxifen or simvastatin. Celecoxib is about 30 times more effective than ibuprofen (Fig. 3 and Table 4). The relative activities of celecoxib, rofecoxib and valdecoxib for the necrotic stage of the actions of NSAIDs, as measured by hemolysis of SRBC, may be associated with an unwanted side reaction. The hemolysis suggests a mechanism proposed much earlier [2, 3] for the effects of NSAIDs on a variety of biological pathways, namely, perturbation of the cell membranes and disruption of normal signaling pathways (Fig. 5). The deesterification of phospholipids could lead to altered signaling. It is not restricted to NSAIDs. Even at high concentrations, AA release is stimulated by tamoxifen. It is independent of estrogen synthesis [9]. The stimulation of AA release by simvastatin is independent of cholesterol synthesis [10]. Simvastatin also hemolyzes SRBC (Fig. 4). The significant correlation between high doses of statins and bone fractures [31] may reflect intercolated cell membranes of cells relevant to bone integrity, i.e. the biological response to high doses of such drugs may depend on the function of the target cell.

Schematic representation of the mechanism of action of high doses of NSAIDs on cell membranes.
COX inhibitors leading to decreased AA metabolism may be the primary pathway for combating pain. The perturbation of the membrane and the accompanying changes in signaling may be responsible for many of their other biological effects, including cancer chemoprevention, reduction of amyloid production in Alzheimer disease [32] and possibly even production of lesions in the gastric mucosa [33]. It would be of interest to assess epidemiologically other conditions that may be affected by exposure to high doses of NSAIDs, e.g., cardiotoxicity. The effects of these agents do not appear to be selective to cancer cells.
The numerous findings of overexpression of COX-2 and elevated PGE2 levels in cancer as well as the genetic evidence [reviewed in [29, 30]] have lead to the concept that COX-2 is a primary target for cancer prevention. Alternatively, the effects on cancer prevention observed with NSAIDs and some other agents may result from the stimulation of AA release followed by the altered downstream signaling. Lypooxygenase (LOX) and LOX products (5-LOX and 12-LOX) are up-regulated in human pancreatic cancer. Inhibitors of LOX induce apoptosis in these human pancreatic cell lines [reviewed in [29]]. Since LOX activity was not examined in the HCT-15 cells, the possibility that LOX is a target for the NSAIDs cannot be ruled out. LOX activity is present in the SRBC undergoing hemolysis but the effect of NSAIDs on the production of LOX products was not studied.
The majority of the compounds tested for release of AA from cells in culture are effective at concentrations of 50 μM or greater. Such concentrations are significantly higher than those found in patients receiving i.e., indomethacin, 20 μM, based on serum levels. However, based only on serum levels, estimations of ligand concentration effective on the target cells can be misleading. For example, sulindac sulfide levels may be 20-fold higher in colonic epithelial cells of guinea pigs than in serum [34]. In addition, comparison of the effects of drug concentrations in cell cultures and in humans is subject to inherent experimental differences. High doses of NSAIDs are used in cell culture experiments while low doses may be effective in man. In cell culture experiments the time of incubation of cells with NSAIDs is of necessity relatively short whereas in patients, the effects are usually observed after much longer periods of time of exposure, sometimes years. The physiological relevance of the high levels of NSAIDs found in vitro has been addressed in recent reviews [29, 30].
Conclusion
A cell that does not express COX activity (HCT-15) releases AA when incubated with NSAIDs and cancer preventive agents. COX negative SRBC also undergo lysis when incubated with NSAIDs, tamoxifen or simvastatin suggesting that the mechanism of action of these drugs involves their intercalation of the membrane and disruption of signaling messages.
Methods
The human colon carcinoma (HCT-15 cell line) and the rat liver (C-9 cell line) were purchased from the American Type Culture Collection (Manassas, VA, USA). They were maintained in Eagle's minimum essential medium (MEM) supplemented with 10% fetal bovine serum. [3H] AA (91.8 Ci/mmol) was obtained from NEN Life Science Products, Inc. (Boston, MA, USA). The washed SRBC were purchased from Lampire Biological Laboratories (Pipersville, PA, USA). All other reagents were from Sigma Chemical Co. (St. Louis, MO, USA) or Calbiochem, (San Diego, CA, USA).
Two days prior to experiments, the HCT-15 or C-9 cells were treated with 0.25% trypsin-EDTA and, after addition of minimal essential media (MEM) containing 10% fetal calf serum, the floating cells were seeded on to 35 mm culture dishes. The plating densities varied from 0.1 to 0.5 × 105 cells/35 mm dish. The freshly seeded cultures were incubated for 24 h to allow for cell attachment. After decantation of MEM containing the fetal bovine serum, 1.0 ml fresh MEM containing 10% fetal bovine serum and [3H] AA (0.2 μCi/ml) was added and the cells incubated for another 24 h. The cells were washed 4 times with MEM and incubated for 19 h with 1.0 ml of MEM containing 1.0 mg BSA/ml (MEM/BSA) and different concentrations of each compound. The culture fluids were then decanted, centrifuged at 2000 x g for 10 min, and 200 μl of the supernate counted for radioactivity. Radioactivity recovered in the washes before the incubation was compared to input radioactivity to calculate the % radioactivity incorporated into the cells. The MEM/BSA values are the control values. The [3H] AA release is presented as a percentage of the radioactivity incorporated by the cells. In 56 experiments, the average release of AA from HCT-15 cells in the presence of MEM/BSA was 3.2 ± 38 %. For all data presented, values are normalized to an MEM/BSA value of 3.2%. Three to five culture dishes were used for each experimental point. The data are expressed as mean values ± SEM. The data were evaluated statistically by the unpaired Student's t-test. A P value < 0.05 was considered significant.
For measuring the effect of NSAIDs on PGI2 production, the C-9 cells were incubated for an additional 24 h with MEM plus 10% fetal bovine serum (minus [3H] AA). The cells were washed three times with MEM and incubated with 5.4 μM lactacystin plus 17 mM TPA in MEM/BSA in the presence of various concentrations of NSAIDs for 6 h. The culture fluids were centrifuged and the supernates analyzed for 6-keto-PGF1α, the stable hydrolytic product of PGI2, by radioimmunoassay [35].
For the hemolysis studies, 100 μl of the SRBC suspension were washed twice with about 15 ml of phosphate buffered saline (pH 7.3) containing BSA (1 mg/ml) (PBS/BSA). The washed cells were resuspended in 50 ml PBS/BSA and 2.5 ml (2.6 × 107/ml) were incubated with varying concentrations of the reagents for 19 h at 37°C. The reaction mixtures were centrifuged and the oxyhaemoglobin measured by absorption at 413 nm. The pellets containing the intact erythrocytes were lysed with 2.5 ml H2O and they too measured for absorption. To determine the number of cells in the experiment, 1 ml of the SRBC suspension was diluted to 15 ml with H2O and the lysate measured for absorption. Controls were performed by incubation of each test reagent in the vehicle alone and measured for absorption in the presence and absence of oxyhaemoglobin. Only sulindac sulfide and indomethacin, at the concentrations used, absorbed at 413 nm. Ibuprofen, at concentrations above 3.5 mM, reduced oxyhaemoglobin absorption.
References
Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971, 231: 232-235.
Abramson SB, Weissmann G: The mechanisms of action of nonsteroidal antiinflammatory drugs. Arthritis Rheum. 1989, 32: 1-9.
Weissmann G: Aspirin. Sci Am. 1991, 264: 84-90.
Kune GA, Kune S, Watson LF: Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48: 4399-4404.
Tashjian AH, Voelkel EF, Goldhaber P, Levine L: Successful treatment of hypercalcemia by indomethacin in mice bearing a prostaglandin-producing fibrosarcoma. Prostaglandins. 1973, 3: 515-524. 10.1016/0090-6980(73)90161-5.
Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, Rigas B: Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol. 1996, 52: 237-245. 10.1016/0006-2952(96)00181-5.
Rigas B, Shiff SJ: Nonsteroidal anti-inflammatory drugs and the induction of apoptosis in colon cells: evidence for PHS-dependent and PHS-independent mechanisms. Apoptosis. 1999, 4: 373-381. 10.1023/A:1009699321946.
Levine L: Does the release of arachidonic acid from cells play a role in cancer chemoprevention?. FASEB J. 2003, 17: 800-802. 10.1096/fj.02-0906hyp.
Levine L: Tamoxifen stimulates arachidonic acid release from rat liver cells by an estrogen receptor-independent, non-genomic mechanism. BMC Cancer. 2003, 3: 24-10.1186/1471-2407-3-24.
Levine L: Statins stimulate arachidonic acid release and prostaglandin I2 production in rat liver cells. Lipids Health Dis. 2003, 2: 1-10.1186/1476-511X-2-1.
Rigas A, Levine L: Arachidonic acid metabolism by rat liver cells (the C-9 cell line). J Pharmacol Exp Ther. 1984, 231: 230-235.
Liu Y, Tonna-DeMasi M, Park E, Schuller-Levis G, Quinn MR: Taurine chloramine inhibits production of nitric oxide and prostaglandin E2 in activated C6 glioma cells by suppressing inducible nitric oxide synthase and cyclooxygenase-2 expression. Brain Res Mol Brain Res. 1998, 59: 189-195. 10.1016/S0169-328X(98)00145-4.
Tomisato W, Tsutsumi S, Hoshino T, Hwang HJ, Mio M, Tsuchiya T, Mizushima T: Role of direct cytotoxic effects of NSAIDs in the induction of gastric lesions. Biochem Pharmacol. 2004, 67: 575-585. 10.1016/j.bcp.2003.09.020.
Kobayashi T, Levine L: Arachidonic acid metabolism by erythrocytes. J Biol Chem. 1983, 258: 9116-9121.
Fitzpatrick F, Liggett W, McGee J, Bunting S, Morton D, Samuelsson B: Metabolism of leukotriene A4 by human erythrocytes. A novel cellular source of leukotriene B4. J Biol Chem. 1984, 259: 11403-11407.
Erman A, Schwartzman M, Raz A: Indomethacin but not aspirin inhibits basal and stimulated lipolysis in rabbit kidney. Prostaglandins. 1980, 20: 689-702. 10.1016/0090-6980(80)90108-2.
Levine L: Tamoxifen and the Rafoxifene analog LY117018: their effects on arachidonic acid release from cells in culture and on prostaglandin I2 production by rat liver cells. BMC Cancer. 2004, 4: 49-10.1186/1471-2407-4-49.
Levine L: Tetrandrine and thapsigargin release arachidonic acid from cells in culture and stimulate prostacyclin production in rat liver cells, but may do so by different pathways. BMC Pharmacol. 2005, 5: 12-10.1186/1471-2210-5-12.
Furuya Y, Lundmo P, Short AD, Gill DL, Isaacs JT: The role of calcium, pH, and cell proliferation in the programmed (apoptotic) death of androgen-independent prostatic cancer cells induced by thapsigargin. Cancer Res. 1994, 54: 6167-6175.
Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT: Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003, 95: 990-1000.
Jiang S, Chow SC, Nicotera P, Orrenius S: Intracellular Ca2+ signals activate apoptosis in thymocytes: studies using the Ca2+-ATPase inhibitor thapsigargin. Exp Cell Res. 1994, 212: 84-92. 10.1006/excr.1994.1121.
Kitamura Y, Miyamura A, Takata K, Inden M, Tsuchiya D, Nakamura K, Taniguchi T: Possible involvement of both endoplasmic reticulum-and mitochondria-dependent pathways in thapsigargin-induced apoptosis in human neuroblastoma SH-SY5Y cells. J Pharmacol Sci. 2003, 92: 228-36. 10.1254/jphs.92.228.
Wang G, Lemos JR, Iadecola C: Herbal alkaloid tetrandrine: fron an ion channel blocker to inhibitor of tumor proliferation. Trends Pharmacol Sci. 2004, 25: 120-123. 10.1016/j.tips.2004.01.009.
Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR: Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A. 1993, 90: 11693-11697.
Wasserman E, Levine L: Quantitative micro-complement fixation and its use in the study of antigenic structure by specific antigen-antibody inhibition. J Immunol. 1961, 87: 290-295.
Liou JY, Aleksic N, Chen SF, Han TJ, Shyue SK, Wu KK: Mitochondrial localization of cyclooxygenase-2 and calcium-independent phospholipase A2 in human cancer cells: implication in apoptosis resistance. Exp Cell Res. 2005, 306: 75-84. 10.1016/j.yexcr.2005.01.011.
Zhang X, Morham SG, Langenbach R, Young DA: Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J Exp Med. 1999, 190: 451-459. 10.1084/jem.190.4.451.
Chan TA, Morin PJ, Vogelstein B, Kinzler KW: Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc Natl Acad Sci U S A. 1998, 95: 681-686. 10.1073/pnas.95.2.681.
Kashfi K, Rigas B: Non-COX-2 targets and cancer: Expanding the molecular target repertoire of chemoprevention. Biochem Pharmacol. 2005, 70: 969-986. 10.1016/j.bcp.2005.05.004.
Rigas B, Kashfi K: Cancer prevention: a new era beyond cyclooxygenase-2. J Pharmacol Exp Ther. 2005, 314: 1-8. 10.1124/jpet.104.080564.
Scranton RE, Young M, Lawler E, Solomon D, Gagnon D, Gaziano JM: Statin use and fracture risk: study of a US veterans population. Arch Intern Med. 2005, 165: 2007-2012. 10.1001/archinte.165.17.2007.
Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH: Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003, 278: 31831-31837. 10.1074/jbc.M303592200.
Tomisato W, Tsutsumi S, Rokutan K, Tsuchiya T, Mizushima T: NSAIDs induce both necrosis and apoptosis in guinea pig gastric mucosal cells in primary culture. Am J Physiol Gastrointest Liver Physiol. 2001, 281: G1092-G1100.
Duggan DE, Hooke KF, Hwang SS: Kinetics of the tissue distributions of sulindac and metabolites. Relevance to sites and rates of bioactivation. Drug Metab Dispos. 1980, 8: 241-246.
Levine L: Measurement of arachidonic acid metabolites by radioimmunoassay. Manual of Clinical Laboratory Immunology. Edited by: Rose NR, Friedman H, Fahey JL. 1986, Washington DC: American Society for Microbiology, 685-691. 3
Acknowledgements
My thanks to Hilda B. Gjika for preparation of the manuscript and to Dr. Armen H. Tashjian, Jr., Department of Genetics and Complex Diseases, Harvard School of Public Health, for his continuing interest in these studies.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Levine, L. Cyclooxygenase expression is not required for release of arachidonic acid from cells by some nonsteroidal anti-inflammatory drugs and cancer preventive agents. BMC Pharmacol 6, 7 (2006). https://doi.org/10.1186/1471-2210-6-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2210-6-7