
Abstract
Background
The receptor for advanced glycation endproducts, RAGE, is involved in the pathogenesis of many inflammatory conditions, which is mostly related to its strong activation of NF-κB but also due to its function as ligand for the β2-integrin Mac-1. To further dissect the stimulus-dependent role of RAGE on leukocyte recruitment during inflammation, we investigated β2-integrin-dependent leukocyte adhesion in RAGE-/- and Icam1-/- mice in different cremaster muscle models of inflammation using intravital microscopy.
Results
We demonstrate that RAGE, but not ICAM-1 substantially contributes to N-formyl-methionyl-leucyl-phenylalanine (fMLP)-induced leukocyte adhesion in TNF-α-pretreated cremaster muscle venules in a Mac-1-dependent manner. In contrast, fMLP-stimulated leukocyte adhesion in unstimulated cremaster muscle venules is independent of RAGE, but dependent on ICAM-1 and its interaction with LFA-1. Furthermore, chemokine CXCL1-stimulated leukocyte adhesion in surgically prepared cremaster muscle venules was independent of RAGE but strongly dependent on ICAM-1 and LFA-1 suggesting a differential and stimulus-dependent regulation of leukocyte adhesion during inflammation in vivo.
Conclusion
Our results demonstrate that RAGE and ICAM-1 differentially regulate leukocyte adhesion in vivo in a stimulus-dependent manner.
Similar content being viewed by others


Background
Leukocyte recruitment into inflamed tissue is considered a fundamental part of the inflammatory process and therefore plays a crucial role in immune defence. Leukocyte recruitment follows a well-defined cascade of events, beginning with the capture of free-flowing leukocytes to the vessel wall, followed by rolling, integrin-mediated firm adhesion to the endothelial layer, postarrest modifications and finally transmigration into tissue [1]. Firm adhesion of leukocytes to the endothelium crucially depends on the β2-integrins LFA-1 (CD11a/CD18, αLβ2) and Mac-1 (CD11b/CD18, αMβ2) which interact with different endothelial ligands such as ICAM-1 and RAGE, the receptor for advanced glycation end products [1–4]. Deficiency of the β2-integrin CD18, an essential part of both β2-integrins LFA-1 and Mac-1, leads to severe recurrent acute and chronic infections in mice and humans, which is caused by impaired leukocyte adhesion [5–7]. Interestingly, deficiency of either LFA-1 or Mac-1 in mice only mildly affects leukocyte recruitment in vivo indicating overlapping functions of LFA-1 and Mac-1 in mediating firm leukocyte adhesion [3, 8]. Similarly, Icam1 knockout mice show only marginal inflammatory defects [9]. Recently, we demonstrated in a model of acute trauma-induced inflammation in the mouse, that the concomitant absence of ICAM-1 and RAGE leads to a dramatic decrease in leukocyte adhesion when compared to control mice or mice where only ICAM-1 or RAGE is absent [3]. These findings provide evidence that the integrin ligands ICAM-1 and RAGE exert overlapping functions [3]. At present it is unclear, if the adhesion molecules RAGE and ICAM-1 cooperate in a similar fashion in other models of inflammation. This prompted us to further dissect the role of ICAM-1 and RAGE for firm leukocyte adhesion under different inflammatory conditions. Using intravital microscopy, we observed leukocyte adhesion in cremaster muscle venules of RAGE and Icam1 knockout mice during trauma- or TNF-α-induced inflammation and additional local stimulation with the leukocyte chemoattractant peptide N-formyl-methionyl-leucyl-phenylalanine (fMLP) or systemic injection of the chemokine CXCL1. Our findings expand previous reports in as much as they show that besides its overlapping function, ICAM-1 and RAGE also exhibit distinct and stimulus-dependent functions in mediating leukocyte adhesion in vivo.
Results
Leukocyte adhesion in trauma-induced inflammation
During trauma-induced inflammation, which is induced by surgical preparation, firm leukocyte arrest is mostly mediated via the β2-integrins LFA-1 and Mac-1 interacting with ICAM-1 and RAGE, respectively [3]. Here, we observed leukocyte adhesion in postcapillary venules of the surgically prepared cremaster muscle of wild type, RAGE-/- and Icam1-/- mice before and during fMLP superfusion. Microvascular and hemodynamic parameters were similar between genotypes and treatment groups and there were no significant changes of systemic leukocyte counts during fMLP superfusion (Table 1). In line with an earlier report [3], leukocyte adhesion in trauma-stimulated cremaster muscle venules was similar in wild type mice, Icam1-/- mice and RAGE-/- mice prior to fMLP superfusion (Figure 1). To explore the contribution of RAGE and ICAM-1 in mediating firm leukocyte adhesion in response to fMLP, we observed leukocyte adhesion in postcapillary venules of the cremaster muscle during 5 min fMLP superfusion. fMLP produced a strong increase in leukocyte adhesion in both RAGE-/- mice and wild type mice. In contrast, fMLP-stimulated leukocyte adhesion was significantly attenuated in Icam1-/- mice suggesting that fMLP-induced leukocyte adhesion in surgically prepared cremaster muscle venules is dependent on ICAM-1, but independent of RAGE (Figure 1).

Leukocyte adhesion in trauma-induced inflammation of cremaster muscle venules during additional fMLP superfusion. Leukocytes adhesion (number of adherent cells/mm2) was observed in trauma-stimulated cremaster muscle venules of wild type (WT) control mice (13 venules in 10 mice), RAGE-/- mice(10 venules in 6 mice) and Icam1-/- mice (4 venules in 4 mice) before and during 5 minutes fMLP-superfusion (10 μM). All values are presented as mean ± SEM and significant differences (p < 0.05) vs. WT control mice are indicated by the asterisk.
Role of Mac-1 and LFA-1 for leukocyte adhesion in fMLP-stimulated trauma-induced inflammation
To further dissect the role of β2-integrins for fMLP-stimulated leukocyte adhesion, we investigated fMLP-induced leukocyte adhesion in exteriorized cremaster muscle venules of wild type and RAGE-/- mice pretreated with blocking mAbs against the β2-integrins Mac-1 or LFA-1 using anti Mac-1 mAb Tib128 (clone M1/70) and anti LFA-1 mAb Tib217 (clone M17/4), respectively. To rule out unspecific effects of the antibodies we injected wild type mice with respective isotype control antibodies. It is important to mention, that we injected all antibodies immediately before fMLP superfusion to identify the role of β2-integrins for fMLP induced leukocyte adhesion only. Hemodynamic and microvascular parameters did not vary significantly between the different groups (Table 1). Before fMLP superfusion leukocyte adhesion was similar in trauma-stimulated cremaster muscle venules of wild type mice and RAGE-/- mice in response to different antibody treatment (Figure 2). This is an expected finding due to the fact that antibodies were injected after induction of trauma. The fMLP-induced increase of leukocyte adhesion (five minutes after fMLP) in wild type or RAGE-/- mice was not affected by pretreatment with Mac-1-blocking mAbTib128. However, fMLP-induced leukocyte adhesion was almost absent in wild type mice or RAGE-/- mice pretreated with LFA-1-blocking mAb Tib217 (Figure 2) suggesting that fMLP-stimulated adhesion is dependent on LFA-1 and ICAM-1 but independent of RAGE and Mac-1.

Role of Mac-1 and LFA-1 for leukocyte adhesion in surgically prepared cremaster muscle venules before and after additional fMLP superfusion. Leukocytes adhesion (number of adherent cells/mm2) is shown before and 5 minutes after fMLP-superfusion (10 μM) in surgically prepared cremaster muscle venules of wild type mice (13 venules in 10 mice) and RAGE-/- mice (10 venules in 6 mice) without antibody treatment and after they were systemically treated with blocking antibodies against murine Mac-1 (Tib128; 19 venules in 6 wild type mice and 14 venules in 4 RAGE-/- mice) or LFA-1 (Tib217, 16 venules in 5 wild type mice and 20 venules in 6 RAGE-/- mice), or their respective isotype control antibodies (IgG2b for Mac-1; 11 venules in 4 wild type mice and IgG2a for LFA-1; 12 venules in 4 wild type mice) immediately before starting fMLP superfusion. All values are presented as mean ± SEM and significant differences (p < 0.05) vs. control mice are indicated by the asterisk.
Leukocyte adhesion in TNF-α-induced inflammation
Leukocyte adhesion in TNF-α-stimulated cremaster muscle venules was observed by intravital microscopy in six wild type mice, five RAGE-/- mice, and four Icam1-/- mice. Hemodynamic and microvascular parameters did not vary significantly between the different groups (Table 2). As described earlier, leukocyte adhesion was significantly reduced in RAGE-/- mice compared to wild type mice or Icam1-/- mice prior to fMLP superfusion (Figure 3). Since fMLP is known to trigger leukocyte adhesion in TNF-α-induced inflammation independent of ICAM-1 [10], we wanted to test if fMLP-induced leukocyte adhesion in TNF-α-treated cremaster muscle venules is dependent on RAGE. Similar to the report by Foy and Ley [10], we observed an increase in leukocyte adhesion during five minutes of fMLP-superfusion in TNF-α-treated cremaster muscle venules of Icam1-/- mice and wild type mice. However, there was no fMLP-induced increase in leukocyte adhesion in RAGE-/- mice (Figure 3), suggesting that leukocyte adhesion is independent of ICAM-1, but dependent on RAGE in this setting.

Leukocyte adhesion in TNF-α-stimulated cremaster muscle venules during additional fMLP superfusion. Leukocytes adhesion (number of adherent cells/mm2) was observed in 3 h TNF-α-stimulated cremaster muscle venules of wild type (WT) control mice (6 venules in 6 mice), RAGE-/- mice (5 venules in 5 mice) and Icam1-/- mice (4 venules in 4 mice) before and during 5 minutes fMLP-superfusion (10 μM). All values are presented as mean ± SEM and significant differences (p < 0.05) vs. WT control mice are indicated by the asterisk.
Role of Mac-1 and LFA-1 for fMLP-induced leukocyte adhesion in TNF-α-stimulated-cremaster muscle venules
To further explore the adhesion molecules involved in fMLP-induced leukocyte adhesion of TNF-α-stimulated cremaster muscle venules, we investigated fMLP-induced leukocyte adhesion in TNF-α-treated cremaster muscle venules using blocking mAbs against Mac-1 and LFA-1. To rule out unspecific effects of the antibodies, we injected wild type mice with respective isotype control antibodies. In parallel to the trauma model, we injected all antibodies immediately before fMLP superfusion to identify the role of β2-integrins for fMLP-induced leukocyte adhesion only. Hemodynamic and microvascular parameters were similar between the different groups (Table 2). As expected in this setting, before fMLP superfusion leukocyte adhesion was similar in TNF-α-stimulated cremaster muscle venules of wild type mice and Icam1-/- mice in the different treatment groups (Figure 4). Five minutes after fMLP superfusion the fMLP-induced increase of leukocyte adhesion was absent in wild type treated with the Mac-1-blocking mAb Tib128 shortly before fMLP superfusion (Figure 4). In contrast, in wild type mice treatment with LFA-1-blocking antibody Tib217 did not alter fMLP-induced leukocyte adhesion compared to wild type mice without antibody treatment, suggesting that fMLP-induced leukocyte adhesion in this setting is mostly dependent on Mac-1 but not LFA-1. Accordingly, treatment of Icam1-/- mice with Mac-1-blocking mAb Tib128 abolished fMLP-triggered leukocyte adhesion, while the absence of ICAM-1 (with or without blockade of LFA-1) did not influence the fMLP-induced increase in leukocyte adhesion (Figure 4). These findings suggest that fMLP-induced leukocyte adhesion in TNF-α-treated cremaster muscle venules is dependent on RAGE and Mac-1, whereas ICAM-1 and LFA-1 are not required.

Role of Mac-1 and LFA-1 for leukocyte adhesion in TNF-α-stimulated cremaster muscle venules before and after additional fMLP superfusion. Leukocytes adhesion (number of adherent cells/mm2) is shown before and 5 minutes after fMLP-superfusion (10 μM) in 3 h TNF-α-stimulated cremaster muscle venules of wild type control mice and Icam1-/- mice, which are systemically treated with blocking antibodies against murine Mac-1 (Tib128; 20 venules in 4 wild type mice and 13 venules in 3 Icam1-/- mice), LFA-1 (Tib217, 13 venules in 4 wild type mice and 14 venules in 3 Icam1-/- mice) or their respective isotype control antibodies (IgG2b for Mac-1; 10 venules in 3 wild type mice and IgG2a for LFA-1; 8 venules in 3 wild type mice) immediately before starting fMLP superfusion or without antibody pretreatment (6 venules in 6 wild type mice and 4 venules in 4 Icam1-/- mice). All values are presented as mean ± SEM and significant differences (p < 0.05) vs. WT control mice and Icam1-/- control mice are indicated by the asterisk.
Leukocyte adhesion following systemic stimulation by chemokine CXCL1 in trauma-induced inflammation
To investigate a potential role of RAGE, ICAM-1 or LFA-1 in chemokine-induced firm leukocyte arrest in vivo, we systemically injected the CXCR2 chemokine CXCL1 (KC) into RAGE-/- and Icam1-/- mice, and into wild type mice with and without anti-LFA-1-blocking or isotype control antibody pretreatment. Systemic injection of CXCL1 has been shown to induce firm leukocyte adhesion in exteriorized cremaster muscle venules of wild type mice [11, 12]. Hemodynamic and microvascular parameters were similar between the different groups (Table 3). Following systemic injection of CXCL1, we observed a significant increase in the number of adherent cells in wild type mice (Figure 5, p < 0.05). Similarly, in RAGE-deficient mice the number of adherent cells significantly increased five minutes after CXCL1 injection (Figure 5, p < 0.05), suggesting that CXCL-1-triggered activation of β2-integrins leading to firm leukocyte arrest does not require RAGE. In contrast, in Icam1-/- mice, systemic injection of CXCL1 did not lead to an increase in leukocyte adhesion (Figure 5) indicating that CXCL1-triggered leukocyte arrest is strongly dependent on ICAM-1. Furthermore, pretreatment of wild type mice with the anti-LFA-1-blocking antibody Tib 217, but not its isotype control antibody, inhibited CXCL1-induced leukocyte arrest, suggesting that LFA-1 and ICAM-1 play a predominant role in mediating CXCL1-triggered leukocyte arrest during trauma-induced inflammation in vivo.

Leukocyte adhesion in CXCL1-stimulated cremaster muscle venules. The number of adherent cells (mean ± SEM) was assessed in exteriorized cremaster muscle venules of RAGE-/- mice (10 venules in 6 mice), Icam1-/- mice (11 venules in 5 mice), untreated (12 venules in 7 mice), anti-LFA-1 antibody (10 venules in 4 mice) or isotype antibody (9 venules in 3 mice) treated wild type mice before and five minutes after systemic injection of CXCL1 (keratinocyte-derived chemokine, KC; 600 ng/mouse). Significant differences (p < 0.05) of the number of adherent leukocytes before CXCL1 vs. after injection of CXCL1 are indicated by *, # and § for wild type mice, RAGE-/- mice and isotype antibody treated wild type mice, respectively.
Stimulus-dependent expression of adhesion molecules in cremaster muscle venules
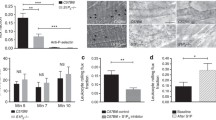
Next, we addressed the question whether the stimulus-dependent role of ICAM-1-LFA vs. RAGE-Mac-1 for leukocyte adhesion is due to altered endothelial expression of adhesion molecules. Immunohistochemistry was used to assess endothelial expression of ICAM-1 and RAGE in postcapillary venules of cremaster muscles obtained from wild type mice either directly postmortem (unstimulated), after exteriorization and superfusion (trauma model) or after 3 h of TNF-α-stimulation followed by surgical preparation (TNF-α model). While we found no RAGE expression on the endothelium of unstimulated cremaster muscle venules of wild type mice (Figure 6A), after trauma and even more pronounced during TNF-α-induced inflammation, RAGE expression could be clearly detected (Figure 6C and 6E, respectively). As illustrated in Figure 6B, D, F, we found endothelial surface expression of ICAM-1 in unstimulated, trauma- and TNF-α stimulated cremaster muscle venules.

Stimulus-dependent expression of RAGE and ICAM-1 in cremaster muscle venules. Immunostaining was conducted to assess endothelial expression of RAGE (A, C, E) and ICAM-1 (B, D, F) in postcapillary venules of cremaster muscles obtained directly postmortem (Unstimulated, upper panel), after exteriorization and 20 min superfusion (Trauma, middle panel), or after 3 h of TNF-α-stimulation and following exteriorization (TNF-α, lower panel; at least 3 mice/group). Application of primary antibody was performed i.v. before harvesting the cremaster muscle in order to stain RAGE and ICAM-1 on the endothelial surface. Biotinylated secondary antibody, peroxidase-conjugated streptavidin and diaminobenzidine (DAB) were used to detect endothelial expression of ICAM-1 and RAGE as brown signal. Counter-staining was performed by Mayer's hemalaun. Reference bar for the representative images is shown in A and represents 20 μm. Intensity of venular anti-RAGE and anti-ICAM-1 immunostaining during trauma- and TNF-α-induced inflammation were analyzed semiquantitatively and presented as mean ± SEM (G; 0 = no, 1 = weak, 2 = medium, 3 = strong signal). Significant differences (p < 0.05) of TNF-α-induced RAGE or ICAM-1 expression vs. trauma are indicated by the asterisk or the pound key, respectively. Relative ratios of expression of RAGE to ICAM-1 are quantified for the trauma and TNF-α model as illustrated.
Beside representative micrographs, the immunostaining were scored semi-quantitatively to compare endothelial expression of RAGE to ICAM-1 between the inflammatory models (Figure 6G). During trauma-induced inflammation we found a moderate expression of RAGE, which was slightly below that of ICAM-1 resulting in a RAGE/ICAM-1 expression ratio of 0.89. TNF-α-stimulation produced a significant upregulation of both RAGE and ICAM-1 compared to trauma-induced inflammation. However, the increase of RAGE expression was stronger than for ICAM-1, which is reflected by a RAGE/ICAM-1 expression ratio of 1.06 indicating that expression of RAGE is more strongly inducible by inflammatory stimulation than expression of ICAM-1.
Stimulus-dependent expression of LFA-1 and Mac-1
To further explore underlying mechanisms, we investigated Mac-1 and LFA-1 expression on isolated wild type neutrophils pretreated with TNF-α, fMLP, or CXCL1 using flow cytometry. LFA-1 expression on wild type neutrophils was strongly upregulated after fMLP and CXCL-1 treatment compared to baseline expression (Figure 7A). However, neutrophils pretreated with TNF-α followed by fMLP treatment only showed a mild increase in LFA-1 expression (Figure 7A). In contrast, expression of Mac-1 was only mildly increased in response to fMLP or CXCL-1, but increased strongly upon stimulation with TNF-α and fMLP when compared to unstimulated neutrophils (Figure 7B). These results demonstrate that LFA-1 and Mac-1 expression are differentially affected by the various stimuli which is in line with the in vivo results shown in Figure 2 and 4.

Stimulus-dependent expression of LFA-1 and Mac-1 on neutrophils. Surface expression of LFA-1 (A) and Mac-1 (B) on bone marrow-derived neutrophils (n = 3 mice) after stimulation with CXCL1 (KC; 120 ng per 106 leukocytes/ml, 15 min), fMLP (10 μM, 106 leukocytes/ml, 15 min), or TNF-α+ fMLP (25 ng TNF-α for 3 h followed by 10 μM fMLP for 15 min in 106 leukocytes/ml) was compared to unstimulated controls (B). Representative histograms are shown from 3 separate experiments.
Discussion
The receptor of advanced glycation endproducts RAGE belongs to the pattern recognition receptors and exerts its central function during acute and chronic inflammations not only as a strong activator of the proinflammatory transcription factor NF-κB but also as endothelial-expressed ligand for the β2-integrin Mac-1 [3, 4, 13]. The biological relevance of RAGE as a ligand for Mac-1 has been recently demonstrated in RAGE-/- × Icam1-/- mice where leukocyte adhesion was almost completely absent in an acute surgically-induced model of inflammation in the cremaster muscle in vivo [3] suggesting that RAGE and ICAM-1 cooperate closely in mediating firm leukocyte adhesion, with RAGE interacting with Mac-1 and ICAM-1 binding to LFA-1. Interestingly, the same study revealed that under circumstances where inflammation was induced by local treatment with TNF-α plus the surgical trauma, leukocyte adhesion was only mildly reduced in the absence of RAGE and ICAM-1 suggesting that alternative, yet unknown integrin ligands exist and that the close cooperation of RAGE and ICAM-1 as β2-integrin ligands is stimulus-dependent (Table 4) [3]. To further investigate the role of RAGE as Mac-1 ligand during leukocyte recruitment, we studied here leukocyte adhesion in cremaster muscles of RAGE-/-, Icam1-/- and wild type mice using different inflammatory models in the cremaster muscle. We show that additional fMLP stimulation during trauma-induced inflammation leads to increased leukocyte adhesion which is independent of RAGE but dependent on ICAM-1, while fMLP superfusion in TNF-α pretreated cremaster muscle venules induces RAGE-dependent adhesion which was independent of ICAM-1 (Table 4). Additional studies using mAbs against Mac-1 and LFA-1 confirmed that RAGE interacts with Mac-1 and ICAM-1 with LFA-1, respectively. These findings imply that the same pro-inflammatory agent (i.e. fMLP) can lead to different biological effects. This is also supported by an earlier report from Foy and Ley [10] showing that fMLP-induced leukocyte adhesion in vivo is only dependent on ICAM-1 in exteriorized cremaster muscles without any other stimulus while additional prestimulation with TNF-α leads to an ICAM-1-independent increase in leukocyte adhesion following fMLP superfusion [10]. The molecular mechanisms responsible for the differential and stimulus-dependent regulation of β2-integrin-mediated leukocyte adhesion are currently unclear.
On the endothelial side, our immunohistochemical findings showed a stimulus-dependent expression of ICAM-1 and RAGE. During trauma-induced inflammation, endothelial RAGE expression was less than ICAM-1 expression, while after TNF-α treatment RAGE expression on the inflamed endothelium was slightly higher than ICAM-1 expression. However, these subtle differences in expression might not explain, why fMLP-induced leukocyte adhesion relies on LFA-1/ICAM-1 during trauma-induced inflammation and on Mac-1/RAGE during TNF-α-stimulated inflammation.
In contrast, our flow cytometric analysis on the expression of Mac-1 and LFA-1 showed that LFA-1 expression on wild type neutrophils was profoundly increased after CXCL1 or fMLP treatment, while pretreatment with TNF-α followed by fMLP did not alter LFA-1 expression. Inversely, Mac-1 upregulation was most prominent on TNF-α plus fMLP treated neutrophils in comparison to CXCL1 or fMLP treatment alone. These findings are in line with our in vivo findings and also to previous observations showing that TNF-α induces Mac-1, but not LFA-1 expression on neutrophils [14, 15].
Besides these more quantitative changes in the expression of Mac-1 and LFA-1 on PMN, additional mechanisms could lie in qualitative changes, i.e. in a stimulus-dependent activation of β2-integrins differing between Mac-1 and LFA-1 [16]. Our group recently showed that ICAM-1 binding to LFA-1 (but not to Mac-1) mediates firm leukocyte adhesion in inflamed cremaster muscle venules, whereas leukocyte crawling in the same setting is dependent on interactions between ICAM-1 and Mac-1 [3]. This suggests that during firm leukocyte arrest, different activation signals are necessary to enable interactions between LFA-1 and ICAM-1 or Mac-1 and ICAM-1. Indeed, different mechanisms of Mac-1 and LFA-1 activation are reported. Fagerholm and colleagues found that ICAM-1 binding to Mac-1 in neutrophils is regulated by α-chain phosphorylation of Mac-1 [17, 18]. As Chatila et al. demonstrated in an earlier study that fMLP treatment of unstimulated PMN did not affect α-chain phosphorylation of Mac-1 [19], this might explain why Mac-1 does not significantly contribute to fMLP-triggered adhesion during trauma-induced inflammation. Noteworthy, LFA-1 and Mac-1 also differ in their binding site on ICAM-1. While LFA-1 uses the D3 domain on ICAM-1 for binding, Mac-1 recognizes the D5 domain on ICAM-1 [20, 21].
Finally, posttranslational glycosylation has been reported as an important regulatory mechanism for leukocyte arrest and integrin-dependent adhesion [22]. For the β2-integrin Mac-1, Feng et al. demonstrated recently that activation of endogenous PMN sialidase leads to desialylation of Mac-1 with a subsequent increase in leukocyte adhesion [23]. The authors explained their findings by a better exposure of the activation epitope on Mac-1 upon desialylation of crucial sialic acid residues on Mac-1.
Conclusions
Our study provides evidence that RAGE is a relevant, but context-dependent Mac-1 ligand during inflammation in vivo. In addition, the presented findings expand our view of the complex and stimulus-dependent regulation of leukocyte adhesion during inflammation (depicted in Table 4), which might lead to new RAGE-based strategies to specifically interfere with the recruitment of immune cells during specific acute and chronic inflammatory conditions.
Methods
Animals
RAGE-/- mice and Icam1-/- mice were generated as described earlier and backcrossed for at least seven generations into the C57BL/6 background [24, 25]. All mice were maintained as breeding colonies at the Central Animal Facility of the University of Heidelberg, Germany. The animal experiments were approved by the Animal Care and Use Committee of the Regierungspräsidium Karlsruhe, Germany (AZ 35-9185.81/G-67/03 and AZ 35-9185.81/G-08/08).
Antibodies and cytokines
In some intravital microscopic experiments, fMLP (10 μM; Sigma, Seisenhofen, Germany) was added to the superfusion buffer to induce additional leukocyte adhesion as described [26]. In certain experiments, recombinant murine TNF-α (R&D) was injected intrascrotally at a dose of 500 ng per mouse 3 hours before intravital microscopy. In some experiments, recombinant murine CXCR2 chemokine CXCL1 (keratinocyte-derived chemokine KC; Peprotech, London, UK) was injected systemically at a dose of 600 ng/mouse. Blocking antibodies against murine Mac-1 (Tib128, clone M1/70, rat IgG2b) and murine LFA-1 (Tib217, clone M17/4, rat IgG2a) were obtained from American Type Culture Collection (ATCC, Manasses, USA) and systemically administered at 100 μg/mouse immediately before starting fMLP superfusion.
Intravital microscopy
Mice were prepared for intravital microscopy as reported recently [12]. Briefly, mice were anesthetized with intraperitoneal (i.p.) injection of ketamine (125 mg/kg body weight, Ketanest®, Pfizer GmBH, Karlsruhe, Germany) and xylazine (12.5 mg/kg body weight; Rompun®, Bayer, Leverkusen, Germany) and placed on a heating pad to maintain body temperature at 37°C. Intravital microscopy was conducted on an upright microscope (Leica; Wetzlar, Germany) with a saline immersion objective (SW40/0.75 numerical aperture, Zeiss, Jena, Germany). To ease breathing mice were intubated using PE 90 tubing (Becton Dickinson and Company, Sparks, MD, USA). The left carotid artery was cannulated with PE 10 tubing (Becton Dickinson and Company, Sparks, MD, USA) for blood sampling and systemic mAb administration.
Cremaster muscle preparation
The surgical preparation of the cremaster muscle for intravital microscopy was performed as previously described [27]. Briefly, the scrotum was opened and the cremaster muscle was mobilized. After longitudinal incision and spreading of the muscle over a cover glass, the epididymis and testis were moved and pinned to the side giving full microscopic access to the cremaster muscle microcirculation. Cremaster muscle venules were recorded via CCD camera (CF8/1; Kappa, Gleichen, Germany) on a Panasonic S-VHS recorder. The cremaster muscle was superfused with thermocontrolled (35°C) bicarbonate-buffered saline. For local treatment with fMLP, 10 μM fMLP was added to the superfusion buffer and administrated over 15 min to the cremaster muscle tissue similar to a previously described protocol [26]. Postcapillary venules under observation were recorded before and during fMLP administration and ranged from 20-40 μm in diameter. Systemic blood samples (10 μl) were taken and assessed for white blood cell count before and after experiment. Blood samples were diluted 1:10 with Türck's solution (Merck, Darmstadt, Germany) and leukocytes concentration was expressed as number of leukocytes per microliter of whole blood using hematocytometer. Microvascular parameters (venular diameter, venular vessel segment length) were measured using an image processing system [28, 29]. Venular centerline red blood cell velocity was measured during the experiment via a dual photodiode and a digital on-line cross-correlation program (Circusoft Instrumentation, Hockessin, USA). An empirical factor of 0.625 was used to convert centerline velocities to mean blood flow velocities [30]. Wall shear rates (γw) were estimated as 4.9 (8vb/d), where vb is mean blood flow velocity and d the diameter of the vessel [31, 32].
Immunohistochemistry
To investigate the endothelial expression of RAGE and ICAM-1 on unstimulated cremaster muscle venules, during trauma- or TNF-α-induced inflammation, we performed immunohistochemical analysis of whole mount cremaster muscles as described [3]. Briefly, primary antibodies against RAGE (polyclonal rat anti mouse; 30 μg/mouse; Santa Cruz, Heidelberg, Germany) or ICAM-1 (YN-1, monoclonal rat anti mouse; 30 μg/mouse; ATCC) were systemically injected and incubated for 10 min ensuring staining of RAGE and ICAM-1 on the endothelial surface. Excess antibody was washed out from the circulation with normal saline solution either before scarifying the mouse (unstimulated) or after 20 min superfusion of the cremaster muscle (trauma- and TNF-α-stimulation). Cremaster muscle whole mounts were surgically prepared as reported previously [3] and transferred onto adhesive slides (Superfrost; Menzel, Germany). Staining of tissue samples for endothelial ICAM-1 and RAGE expression was performed using diaminobenzidine (DAB kit; Vector Lab, Burlingame, USA). Analysis of stained slides was conducted semiquantitatively in a blinded manner (0 = no, 1 = weak, 2 = medium, 3 = strong signal) on a Leica DMRB upright microscope and a ×25/0,75 NA oil immersion objective (both Leica, Wetzlar, Germany). Photographs of the samples were taken using a color CCD camera (KAPPA).
Flow cytometry
The expression of Mac-1 and LFA-1 on bone marrow-derived neutrophils was assessed by flow cytometry as described previously [3]. After red blood cell lysis, 106 leukocytes/ml were stimulated for 15 min with 120 ng CXCL1 (KC) or 10 μM fMLP alone, or with 25 ng TNF-α over 3 h followed by 10 μM fMLP for 15 min (all at 37°C). Next, cells were incubated in the dark with FITC-conjugated anti- Mac-1 mAb M1/70 (1 μg/105 cells, rat IgG2b; eBioscience, San Diego, USA), FITC-conjugated LFA-1 mAb M17/4 (1 μg/105 cells, rat IgG2a; eBioscience, San Diego, USA) or respective FITC-conjugated isotype control antibodies (1 μg/105 cells, rat IgG2b or rat IgG2a; eBioscience, San Diego, USA) to detect anti-Mac-1 and LFA-1 signals, respectively. Mac-1 and LFA-1 expression was assessed on 10.000 cells/mouse within the neutrophil cluster defined by forward-side scatter analysis using LSRII with DIVA software package (Becton Dickinson, San Jose, USA). Expression of Mac-1 and LFA-1 upon stimulation with fMLP, CXCL1, or TNF-α followed by fMLP was compared to unstimulated cells and their respective isotype controls.
Statistics
Sigma Stat 3.5 (Systat Software, Erkrath, Germany) was used for statistical analysis. Leukocyte counts, vessel diameters, centerline velocity, leukocyte adhesion and wall shear rates were compared with one-way ANOVA followed by a multiple pairwise comparison test (Dunn's test) or by Wilcoxon rank-sum test, as appropriate. Statistical significance was set at p < 0.05.
Abbreviations
- fMLP:
-
N-formyl-methionyl-leucyl-phenylalanine
- ICAM-1:
-
intercellular adhesion molecule-1
- KC:
-
keratinocyte-derived chemokine (CXCL1)
- LFA-1:
-
leukocyte functional antigen-1
- Mac-1:
-
macrophage antigen complex 1
- NF-kB:
-
Nuclear Factor kB
- RAGE:
-
receptor for advanced glycation endproducts
- TNF-α:
-
tumor necrosis factor-α.
References
Ley K, Laudanna C, Cybulsky MI, Nourshargh S: Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007, 7: 678-689. 10.1038/nri2156.
Huang MT, Larbi KY, Scheiermann C, Woodfin A, Gerwin N, Haskard DO, Nourshargh S: ICAM-2 mediates neutrophil transmigration in vivo: evidence for stimulus specificity and a role in PECAM-1-independent transmigration. Blood. 2006, 107: 4721-4727. 10.1182/blood-2005-11-4683.
Frommhold D, Kamphues A, Hepper I, Pruenster M, Lukic IK, Socher I, Zablotskaya V, Buschmann K, Lange-Sperandio B, Schymeinsky J, et al: RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010, 116: 841-849. 10.1182/blood-2009-09-244293.
Bierhaus A, Nawroth PP: Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia. 2009, 52: 2251-2263. 10.1007/s00125-009-1458-9.
Anderson DC, Springer TA: Leukocyte adhesion deficiency: An inherited defect in the Mac-1, LFA-1 and p150,95 glycoproteins. Annu Rev Med. 1987, 38: 175-194. 10.1146/annurev.me.38.020187.001135.
Scharffetter-Kochanek K, Lu HF, Norman K, Vannood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, et al: Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998, 188: 119-131. 10.1084/jem.188.1.119.
Kishimoto TK, Baldwin ET, Anderson DC: Inflammation: Basic Principles and Clinical Correlates. Edited by: Gallin JI, Snyderman R. 1999, Philadelphia: Lippincott Williams and Wilkins, 537-570. Volume 3rd
Dunne JL, Ballantyne CM, Beaudet AL, Ley K: Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002, 99: 336-341. 10.1182/blood.V99.1.336.
Sligh JE, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL: Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1993, 90: 8529-8533. 10.1073/pnas.90.18.8529.
Foy DS, Ley K: Intercellular adhesion molecule-1 is required for chemoattractant-induced leukocyte adhesion in resting, but not inflamed, venules in vivo. Microvasc Res. 2000, 60: 249-260. 10.1006/mvre.2000.2272.
Smith ML, Olson TS, Ley K: CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004, 200: 935-939. 10.1084/jem.20040424.
Frommhold D, Ludwig A, Bixel MG, Zarbock A, Babushkina I, Weissinger M, Cauwenberghs S, Ellies LG, Marth JD, Beck-Sickinger AG, et al: Sialyltransferase ST3Gal-IV controls CXCR2-mediated firm leukocyte arrest during inflammation. J Exp Med. 2008, 205: 1435-1446. 10.1084/jem.20070846.
Chavakis T, Bierhaus A, Al Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP: The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003, 198: 1507-1515. 10.1084/jem.20030800.
Miyata R, Iwabuchi K, Watanabe S, Sato N, Nagaoka I: Short exposure of intestinal epithelial cells to TNF-alpha and histamine induces Mac-1-mediated neutrophil adhesion independent of protein synthesis. J Leukoc Biol. 1999, 66: 437-446.
Pichyangkul S, Schick D, Schober W, Dixon G, Khan A: Increased expression of adhesive proteins on leukocytes by TNF alpha. Exp Hematol. 1988, 16: 588-593.
Kim M, Carman CV, Yang W, Salas A, Springer TA: The primacy of affinity over clustering in regulation of adhesiveness of the integrin {alpha}L{beta}2. J Cell Biol. 2004, 167: 1241-1253. 10.1083/jcb.200404160.
Fagerholm SC, Varis M, Stefanidakis M, Hilden TJ, Gahmberg CG: alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood. 2006, 108: 3379-3386. 10.1182/blood-2006-03-013557.
Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Gronholm M: Regulation of integrin activity and signalling. Biochim Biophys Acta. 2009, 1790: 431-444.
Chatila TA, Geha RS, Arnaout MA: Constitutive and stimulus-induced phosphorylation of CD11/CD18 leukocyte adhesion molecules. J Cell Biol. 1989, 109: 3435-3444. 10.1083/jcb.109.6.3435.
Diamond MS, Staunton DE, Marlin SD, Springer TA: Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991, 65: 961-971. 10.1016/0092-8674(91)90548-D.
Luo BH, Carman CV, Springer TA: Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007, 25: 619-647. 10.1146/annurev.immunol.25.022106.141618.
Sperandio M, Gleissner CA, Ley K: Glycosylation in immune cell trafficking. Immunol Rev. 2009, 230: 97-113. 10.1111/j.1600-065X.2009.00795.x.
Feng C, Zhang L, Almulki L, Faez S, Whitford M, Hafezi-Moghadam A, Cross AS: Endogenous PMN sialidase activity exposes activation epitope on CD11b/CD18 which enhances its binding interaction with ICAM-1. J Leukoc Biol. 2011, 90: 313-321. 10.1189/jlb.1210708.
Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, et al: Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004, 113: 1641-1650.
Xu H, Gonzalo JA, St. Pierre Y, Williams IR, Kupper TS, Cotran RS, Springer TA, Gutierrez-Ramos JC: Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994, 180: 95-109. 10.1084/jem.180.1.95.
Frommhold D, Mannigel I, Schymeinsky J, Mocsai A, Poeschl J, Walzog B, Sperandio M: Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo. BMC Immunol. 2007, 8: 31-10.1186/1471-2172-8-31.
Sperandio M, Thatte A, Foy D, Ellies LG, Marth JD, Ley K: Severe impairment of leukocyte rolling in venules of core 2 glucosaminyltransferase-deficient mice. Blood. 2001, 97: 3812-3819. 10.1182/blood.V97.12.3812.
Klyscz T, Junger M, Jung F, Zeintl H: [Cap image--a new kind of computer-assisted video image analysis system for dynamic capillary microscopy]. Biomed Tech (Berl). 1997, 42: 168-175. 10.1515/bmte.1997.42.6.168.
Zeintl H, Sack FU, Intaglietta M, Messmer K: Computer assisted leukocyte adhesion measurement in intravital microscopy. Int J Microcirc Clin Exp. 1989, 8: 293-302.
Lipowsky HH, Zweifach BW: Application of the "two-slit" photometric technique to the measurement of microvascular volumetric flow rates. Microvasc Res. 1978, 15: 93-101. 10.1016/0026-2862(78)90009-2.
Smith ML, Long DS, Damiano ER, Ley K: Near-wall micro-PIV reveals a hydrodynamically relevant endothelial surface layer in venules in vivo. Biophys J. 2003, 85: 637-645. 10.1016/S0006-3495(03)74507-X.
Long DS, Smith ML, Pries AR, Ley K, Damiano ER: Microviscometry reveals reduced blood viscosity and altered shear rate and shear stress profiles in microvessels after hemodilution. Proc Natl Acad Sci USA. 2004, 101: 10060-10065. 10.1073/pnas.0402937101.
Acknowledgements
We thank Melitta Weissinger, Inna Babushkina and Natascha Braach for their excellent technical assistance in performing intravital microscopy and immunohistochemistry and Axel Erhardt for his expert daily mice care. Finally, we thank Britta Engelhardt, Bern, Switzerland, for providing Icam1-/- mice. This work was in part supported by grants from the Deutsche Forschungsgemeinschaft (SFB938 to AB) and LMU Innovativ BioImaging (MS).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DF designed research, performed research, analyzed data and wrote the manuscript, AK performed research, analyzed data and prepared the manuscript. KB, SD, RT, VZ and BLS carried out research and analyzed data, PPN contributed analytical tools, JP contributed analytical tools, AB provided RAGE-/- and Icam1-/- mice and edited the manuscript, MS designed research and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Frommhold, D., Kamphues, A., Dannenberg, S. et al. RAGE and ICAM-1 differentially control leukocyte recruitment during acute inflammation in a stimulus-dependent manner. BMC Immunol 12, 56 (2011). https://doi.org/10.1186/1471-2172-12-56
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2172-12-56