
Abstract
Background
The annual fish Nothobranchius furzeri is characterized by a natural dichromatism with yellow-tailed and red-tailed male individuals. These differences are due to different distributions of xanthophores and erythrophores in the two morphs. Previous crossing studies have showed that dichromatism in N. furzeri is inherited as a simple Mendelian trait with the yellow morph dominant over the red morph. The causative genetic variation was mapped by linkage analysis in a chromosome region containing the Mc1r locus. However, subsequent mapping showed that Mc1r is most likely not responsible for the color difference in N. furzeri. To gain further insight into the molecular basis of this phenotype, we performed RNA-seq on F2 progeny of a cross between N. furzeri male and N. kadleci female.
Results
We identified 210 differentially-expressed genes between yellow and red fin samples. Functional annotation analysis revealed that genes with higher transcript levels in the yellow morph are enriched for the melanin synthesis pathway indicating that xanthophores are more similar to melanophores than are the erythrophores. Genes with higher expression levels in red-tails included xanthine dehydrogenase (Xdh), coding for a biosynthetic enzyme in the pteridine synthesis pathway, and genes related to muscle contraction. Comparison of DEGs obtained in this study with genes associated with pigmentation in the Midas cichlid (A. citrinellus) reveal similarities like involvement of the melanin biosynthesis pathway, the genes Ptgir, Rasef (RAS and EF-hand domain containing), as well as genes primarily expressed in muscle such as Ttn and Ttnb (titin, titin b).
Conclusions
Regulation of genes in the melanin synthetic pathway is an expected finding and shows that N. furzeri is a genetically-tractable species for studying the genetic basis of natural phenotypic variations. The current list of differentially-expressed genes can be compared with the results of fine-mapping, to reveal the genetic architecture of this natural phenotype. However, an evolutionarily-conserved role of muscle-related genes in tail fin pigmentation is novel finding and interesting perspective for the future.
Similar content being viewed by others


Background
Pigmentation is a tractable phenotypic trait that can be easily quantified, and shows remarkable inter- and intra-specific variation. Differences in pigmentation are the most apparent trait that often distinguishes natural populations of a species and/or closely-related species [1–4]. Pigmentation has received considerable attention both in the context of speciation and adaptation [5–10]. Therefore, pigmentation represents a manageable case for studying molecular mechanisms underlying evolutionary processes. Much effort was invested in recent years to identify genetic pathways that determine pigmentation variations in humans and in natural animal populations that revealed a substantial conservation in the pathways controlling pigmentation in vertebrates [2, 8, 11, 12], with several genes associated with variations in mammal pigmentation that share the same function in teleost fish, such as Kit, Mc1r, Oca2, Scl24a, Sox10, Mitf, and Ednrb[8, 11].
Teleost fish are characterized by the existence of further pigmentation cells in addition to melanocytes (melanophores in fish). These are xanthophores (ochre or yellow), erythrophores (red), leucophores (whitish), iridophores (metallic or iridescent) and cyanophores (blue) [13]. Orange to red pigmentation can originate from carotenoids and broadly discussed as a marker of phenotypic quality or dietary history or endogenous pteridines [14–16]. A very common color variant in captive ornamental teleost species is the so-called "gold" or "orange" variant in which the melanophore density is reduced and the xanthophores become dominant giving the animals a yellow-orange color. A similar natural pattern is observed in Lake Malawi cichlids and is controlled by regulatory elements in the Pax7 locus [4]. In Midas cichlids, pigmentation may change in the course of ontogeny giving rise to the gold morph, and a recent transcriptomic study identified genes regulated during this transition [2].
The African teleost Nothobranchius furzeri is the shortest-lived vertebrate that can be cultured in captivity and was recently introduced as a model organism in the context of aging research. A reference transcriptome for N. furzeri was recently generated [17]. N. furzeri is characterized by a striking dichromatism with two phenotypes: a "yellow-tail" morph that shows a yellow submarginal band and a black marginal band (Figure 1A), and a "red-tail" morph with a broad red marginal band. These two morphs coexist over a broad portion of the distribution range in Southern Mozambique, with the yellow morph predominating in the margin of the distribution range [18]. The two morphs are not genetically differentiated implying the absence of assortative mating [19, 20]. The sister taxon to N. furzeri is N. kadleci[20], with parapatric distribution and showing a solid red tail. Previous crossing studies have revealed that dichromatism in N. furzeri is inherited as a simple Mendelian trait with the yellow morph dominant over the red morph [21]. The causative genetic variation was mapped by linkage analysis in a chromosome region containing the Mc1r locus, but subsequent mapping of an Mc1r marker showed that the gene is most likely not responsible for the color difference in N. furzeri[20, 21].

Tail dichromatism in Nothobranchius fish. A: The yellow morph of N. furzeri has a spotted tail with a sub-marginal yellow band and a marginal black band [Photo by Alexander Dorn]. N. kadleci have a solid red tail in general and sometimes with bluish spots in the proximal region. B: Corresponding yellow and red tail portions were clipped for RNA isolation. C: The yellow band of N. furzeri contains the yellow xanthophores almost exclusively, whereas the corresponding portions in N. kadleci have the black melanophores and red erythrophores (magnification, 10x). D: Hierarchical clustering of expression profiles derived from four RNA-seq pools of yellow (Y1 – Y4) and four pools of red (R1 –R4) tail samples. The profiles of samples correspond with the yellow and red tail phenotypic categories.
In the present paper, we used RNA-seq to identify genes differentially expressed in the yellow vs. red morphs of N. furzeri and N. kadleci.
Results and discussion
Tail expression profiles correspond with dichromatism
To make sure that genetic mechanisms of tail pigmentation are identical in the two species, we performed a complementation test by crossing individuals of the MZM-0703 red-tailed strain of N. furzeri[22] with those of N. kadleci in both possible combinations of parents. All 24 F1 male offspring from the two crosses showed a red tail supporting the assumption that the same locus is associated with red colour in both species.
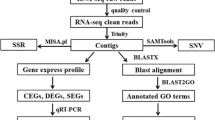
In order to identify differences in gene expression between the two colour morphs that are independent of the genetic background, we used a F2 hybrid panel obtained by crossing a yellow-tailed male of the N. furzeri strain GRZ with a female N. kadleci. A clear segregation of the two phenotypes was observed in F2 generation (Ng’oma, unpublished data) confirming the Mendelian inheritance of this trait [21]. At the age of 16 weeks, the yellow submarginal band of the caudal fin and the corresponding region of red-tailed fish were excised taking care of excluding the black marginal band (Figure 1B,C). Four RNA pools of each colour morph (each pool containing samples of four fish) were analyzed by Illumina RNA-seq resulting in 334,588,906 reads (163,622,787 for red sample pools and 170,966,119 for yellow sample pools) having an average of around 42 million reads per sample pool. About 55% of the reads could be mapped to the reference transcriptome of N. furzeri. Hierarchical clustering of the reads showed that the samples group in correspondence with the yellow and red phenotypic categories (Figure 1D). In total, 210 differentially-expressed genes (DEGs) were identified by the intersection of the statistical tests in the packages edgeR and DEseq (see also Methods). Of the 210 DEGs, 119 were up regulated, while 91 were down-regulated in the yellow samples with respect to the red samples. A full list of DEGs is provided in Additional file 1. Project data are deposited in the Short Reads Archive (SRA), reference number SRP034010.
In order to confirm the expression changes detected by RNA-seq we performed qPCR for four sample genes selected on the basis of fold change (log2 fold change yellow vs. red morph; P value of adjusted meanof DESeq and edgeR) or known role in pigmentation biology: Esrrga (5.66; P = 6.74×10-19), Tyr (2.51; P = 3:29×10-08), Pax3 (-2.82; P = 1.75×10-16), and Sox10 (0.67; P = 5.29×10-3). Estrogen-related receptor gamma a (Esrrga) was the gene with highest fold change in yellow samples, Tyrosinase (Tyr) is a rate-limiting biosynthetic enzyme of melanin, and the transcription factors Pax3 and Sox10 are important in the differentiation of melanocytes [11, 23]. In all four cases, the direction of change was consistent between the two methods (Figure 2). Thus, as already shown [24], we found coherence between RNA-seq and qPCR.

RT-qPCR validation of sample genes regulated in RNA-seq data. Four sample genes were selected based on fold change or known role in melanocyte biology (log2 fold change yellow vs. red morph). The same individuals whose expression was quantified with RNA-seq were used in qPCR. Expression was normalized to TBP, as internal control; error bars represent standard deviation.
Melanogenesis is the main process regulated between morphs
To assess which biological processes may be modulated by the DEGs, Gene Ontology (GO) analysis was performed. Lists of genes with significantly different transcript levels were analyzed with the Functional Annotation Clustering (FAC) tool implemented in the Database for Annotation, Visualization and Integrated Discovery (DAVID) [25]. FAC allows clustering of GO categories sharing significant amount of genes and partially obviates to redundancy of GO classes by providing a more compact output than conventional GO enrichment analysis. The FAC analysis of 119 higher expressed genes resulted into 19 enriched functional clusters under the high stringency option. FAC1 (pigmentation and melanogenesis) was the most biologically important gene group with enrichment score of 3.54 (Figure 3A). Melanin biosynthetic process (GO: 0042438) was the most significant biological process (P = 7.9E×10-06) in yellow tails (see Additional file 2). FAC analysis of the 91 lower expressed genes yielded 12 enriched functional gene groups. Muscle components and organization were the most significant biological processes, such as Z disc (P 0 7.6E-04), myofibril (P = 9.1E-04), and contractile fibre part (P = 9.8E-04) (Figure 3B; Additional file 2).

Gene Ontology. DAVID Functional Annotation Clustering (FAC) analysis of differentially expressed genes (DEGs) obtained by comparison of yellow vs. red tail RNA samples. A. Enriched functional gene clusters for the 119 up-regulated genes. B. Enriched functional gene clusters for the 91 down-regulated genes. Significance is determined by corresponding enrichment scores.
FAC1 revealed five pigmentation genes (Dct, Tyrp1, Tyr, Oca2, and Sox10) with fold-change range of 1.8 – 7.9 (Additional file 2). Tyr, Tyrp1 and Dct are members of the tyrosinase gene family that arose by ancient duplications and catalyse conversion of amino acid tyrosine to melanin[26]. Deletions in Oca2 were shown to be responsible for parallel evolution of albinism in the Mexican cave fish [10]. Sox10 in together with Mitf regulate melanocyte differentiation by activating Tyr[27, 28]. The remaining FAC clusters of up-regulated genes were dominated by solute carrier and channel genes including Slc24a5, presumably a cation exchanger linked with variations in number, size and density of melanosomes in humans and zebrafish [29]. Muscle-related processes featured prominently in the FAC clusters with lower transcript levels (Additional file 2). Cluster 3 included Xdh (xanthine dehydrogenase), 2.1-fold lower expressed which is a key enzyme in the synthesis of pteridine pigments. This gene group also contained solute carrier protein genes including Ano8 (anoctamin 8). Under relaxed clustering stringency, Mreg (melanoregulin) was also observed.
The enrichment of melanogenesis-related terms with respect to higher expression in yellow fish suggests that melanocytes are found more often in yellow- with respect to red-tailed fish. However, tail micrographs (Figure 1C) surprisingly showed that the yellow submarginal band actually lacked melanocytes, which were abundant in corresponding red tail regions. This suggests that the xanthophores were connected with melanogenesis, thus indicating that xanthophores are functionally more related to the melanocytes and most likely ontogenetically closer to the melanocyte lineage than to the erythrophores. Indeed, studies have reported on the role of Pax3/Pax7 fate switch to myogenesis [30] in which pro-myogenesis Hedgehog and Fgf8 signalling suppress Pax3/Pax7, and promote myogenic regulatory factors. A further study on the role of Pax3 and Pax7 genes in the zebrafish neural crest [31] demonstrated sequential actions of Pax3 and Pax7 in fate switch induction from xanthophores to melanophores in Pax3 knock-down embryos. Higher expression of Xdh in the red-tail samples indicates that the red pigment is pteridine-based. Both sepiapterin and drosopterin are formed from 6-pyruvoyl-H4pterin, and Xdh is required for the formation of yellow pigments from sepiapterin [11]. However, the pathway leading to the formation of the orange-red pigments from drosopterin is yet to be elucidated. The regulation of Xdh in this study may suggest a possible role of this gene in Nothobranchius tail pigmentation.
Regulation of muscle genes in Midas cichlids
Regulation of genes in the melanin synthetic pathway is an expected finding. A surprising observation is the regulation of muscle-related genes. The tail is a heterogeneous source of RNA, containing various other cells (bone, cartilage, etc.) in addition to pigment cells. We compared observed differences between Nothobranchius colour morphs with expression changes observed in the Midas cichlids in relation to transition from the grey to the gold morph [2] and identified 9 genes with consistent expression changes in the two species (Table 1). It is interesting that just a handful of muscle genes were regulated, and that muscle genes were regulated in both Nothobranchius and Midas.
We noted above the role of known colour genes (Pax3/Pax7) in muscle development. Pax3 plays key roles in melanocyte development (proliferation, survival and migration), and melanoma by maintaining a less differentiated ‘stem’ cell-like phenotype [32]. Further, a cis-regulatory mutation in Pax7 drives the differentiation of the orange-blotch phenotype from the common male nuptial colouration in Lake Malawi cichlids [4]. Similarly, the intensity of coat colour is reduced in mice lacking myosine heavy chain genes such as Myo5a (or Myha) due to loss of connections between melanosomes and F-actin which affect melanosome distribution in the cell [33, 34]. It would be worthwhile to analyze gene expression at the embryonic stage when cell fate specification and differentiation is still underway. The challenge though would be in distinguishing embryo colour morphs given that the manner and timing in which erythrophores and xanthophores differentiate from their precursor is not understood. Taken together, these observations and our results suggest a possible link between the pigmentation and mayogenic pathways in Nothobranchius. Presently, the functional significance of these genes for red pigmentation remains unknown and forms an interesting perspective for the future.
Conclusions
This study successfully identified pigmentation as the major biological process altered between tail colour morphs consistent with knowledge in other species. Our results therefore show that N. furzeri is a genetically-tractable species for studying the genetic basis of natural phenotypic variations, not only through classical linkage methods as in [21], but also amenable to modern hi-tech genomic approaches. Thus, the current list of differentially-expressed genes can feed back to, and be compared with the results of fine-mapping of the colour locus that is ongoing, to reveal the genetic architecture of this natural phenotype.
Methods
Fish breeding and sample preparation
A male of the yellow tail morph, N. furzeri (GRZ strain) was mated to a female of the red tail morph, N. kadleci resulting into 34 F1 fish. From sib mating of several F1 pairs, 16 yellow tailed and 16 red tailed F2 fish were sacrificed at 16 weeks of age for transcriptome comparison. Care and experimentation with fish followed protocols approved by the local authority in the State of Thüringia (Veterinaer- und Lebensmittelueberwachungsamt). Yellow sub-marginal strips of the tailfin and corresponding regions of red-tailed fish were excised (Figure 1B) and transferred to 1.5 ml tubes containing 600 μl RNAlater (Qiagen, Hilden, Germany) and stored at -20°C until RNA isolation.
RNA sequencing
Total RNA was isolated using QIAzol (Qiagen) following manufacturer’s protocol with modifications as described elsewhere [35]. Four pools, each containing 1 μg RNA, were prepared for each phenotype by combining RNA from four fish in equimolar amounts. RNA samples were checked for quality using an Agilent Bioanalyzer 2100 in combination with the Agilent RNA 6000 Nano Kit (Agilent Technologies) to have a RIN value > 7. Libraries were prepared using Illumina’s TruSeq™ RNA Sample Prep Kit v2 (Illumina Inc.) following the manufacturer’s description, and sequenced with Illumina HiSeq 2000 in 50 bp single-read mode.
Identification of differentially expressed genes
The resulting reads were mapped to nonredundant set of transcript contigs representing the longest transcript contig per gene of N. furzeri[17] using ELAND (Illumina Inc.). Based on the mapping results the reads per transcript were counted. The count data were used for determination of differentially expressed genes (DEGs) using the packages edgeR [36] and DESeq [37] within the statistical environment R (R Development Core Team, 2012). The p-values were adjusted for multiple testing using the Benjamini-Hochberg algorithm [38]. Genes were regarded to be differentially expressed when both tests showed a significance ≤ 0.01. This resulted in a total number of 210 genes which were found to be differentially expressed.
Gene ontology analysis
Hierarchical clustering was performed to sort samples and genes with similar profiles using a dedicated R-script, count_genes_per_transcript.pl [39]. Gene Ontology analysis to identify biological processes likely modulated by the DEGs was performed in DAVID tools [40]. All analysis in DAVID was performed using the Functional Annotation Clustering (FAC) module set to high stringency. The FAC enrichment score (-log10P-values/n) for each cluster was graphed. The enrichment score gives indication of the biological significance of the clusters [41]. Tail micrographs were captured from corresponding yellow and red sub-marginal regions at 10× using the AxioVision Product Suite (Carl Zeiss, Jena, Germany).
Validation by RT-qPCR
cDNA was synthesized in 20 μl volumes for each sequenced individual’s RNA using the QuantiTect® Reverse Transcription Kit (Qiagen) following the supplier’s protocol, and diluted with 200 μl ultrapure water. PCR primers for a sample of genes regulated in RNA-seq including Esrrga, Tyr, Sox10, and Pax3 were designed from CDS across introns (forward and reverse): Esrrga - GAGGATAGGGAAGAGAAG and AACAGAGAGCAGTGGACG; Tyr- GCTCTGTCTTCTCTTCTTG and ATGTTGGCGGTGCGGTCC; Sox10 - ATCAGACGACGAAGAGGAG and GCAGGTGGGGGTGTTGG; Pax3 - GGGAAGGAGGCTGGATAG and CGTGGGTAGTTCTGGTGAG. The tatabox binding protein (Tbp– CGGTTGGAGGGTTTAGTCCT and GCAAGACGATTCTGGGTTTG) was used for normalization. Real-time PCR was performed with the RotorGene 6000 (Qiagen) GoTaq® qPCR Master Mix (Promega) using 4 ng cDNA per reaction in triplicate on the following temperature profile: 95°C for 2 min; 35× (95°C, 30 s, 60°C, 30 s, 72°C, 25 s). Statistical analysis of RT-qPCR results was conducted with the relative expression software tool, REST (Qiagen) [42], which implements a mathematical model comparing treatment and control samples and significance is tested by a randomization test.
References
Elmer KR, Lehtonen TK, Meyer A: Color assortative mating contributes to sympatric divergence of neotropical cichlid fish. Evolution. 2009, 63 (10): 2750-2757. 10.1111/j.1558-5646.2009.00736.x.
Henning F, Jones J, Franchini P, Meyer A: Transcriptomics of morphological color change in polychromatic Midas cichlids. BMC Genomics. 2013, 14 (1): 171-10.1186/1471-2164-14-171.
Hollocher H, Hatcher JL, Dyreson EG: Evolution of abdominal pigmentation differences across species in the Drosophila dunni subgroup. Evolution. 2000, 54 (6): 2046-2056. 10.1111/j.0014-3820.2000.tb01248.x.
Roberts RB, Ser JR, Kocher TD: Sexual conflict resolved by invasion of a novel sex determiner in Lake Malawi cichlid fishes. Science (New York, NY). 2009, 326 (5955): 998-1001. 10.1126/science.1174705.
Greenwood AK, Cech JN, Peichel CL: Molecular and developmental contributions to divergent pigment patterns in marine and freshwater sticklebacks. Evol Dev. 2012, 14 (4): 351-362. 10.1111/j.1525-142X.2012.00553.x.
Gross JB, Borowsky R, Tabin CJ: A novel role for Mc1r in the parallel evolution of depigmentation in independent populations of the cavefish Astyanax mexicanus. PLoS Genet. 2009, 5 (1): e1000326-10.1371/journal.pgen.1000326.
Hoekstra HE: Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity. 2006, 97: 222-234. 10.1038/sj.hdy.6800861.
Hubbard JK, Uy JA, Hauber ME, Hoekstra HE, Safran RJ: Vertebrate pigmentation: from underlying genes to adaptive function. Trends Genet. 2010, 26 (5): 231-239. 10.1016/j.tig.2010.02.002.
O'Quin CT, Drilea AC, Roberts RB, Kocher TD: A Small Number of Genes Underlie Male Pigmentation Traits in Lake Malawi Cichlid Fishes. J Exp Zool B Mol Dev Evol. 2012, 318 (3): 199-208. 10.1002/jez.b.22006.
Protas ME, Hersey C, Kochanek D, Zhou Y, Wilkens H, Jeffery WR, Zon LI, Borowsky R, Tabin CJ: Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat Genet. 2006, 38 (1): 107-111. 10.1038/ng1700.
Braasch I, Schartl M, Volff JN: Evolution of pigment synthesis pathways by gene and genome duplication in fish. BMC Evol Biol. 2007, 7: 74-10.1186/1471-2148-7-74.
Cheng KC: Skin color in fish and humans: impacts on science and society. Zebrafish. 2008, 5 (4): 237-242. 10.1089/zeb.2008.0577.
Fujii R: The regulation of motile activity in fish chromatophores. Pigment Cell Res. 2000, 13 (5): 300-319. 10.1034/j.1600-0749.2000.130502.x.
Baube CL: Manipulations of signalling environment affect male competitive success in three-spined sticklebacks. Anim Behav. 1997, 53 (4): 819-833. 10.1006/anbe.1996.0347.
Grether GF, Hudon J, Endler JA: Carotenoid scarcity, synthetic pteridine pigments and the evolution of sexual coloration in guppies (Poecilia reticulata). Proc R Soc B. 2001, 268 (1473): 1245-1253. 10.1098/rspb.2001.1624.
Theis A, Salzburger W, Egger B: The Function of Anal Fin Egg-Spots in the Cichlid Fish Astatotilapia burtoni. PLoS One. 2012, 7 (1): e29878-10.1371/journal.pone.0029878.
Petzold A, Reichwald K, Groth M, Taudien S, Hartmann N, Priebe S, Shagin D, Englert C, Platzer M: The transcript catalogue of the short-lived fish Nothobranchius furzeri provides insights into age-dependent changes of mRNA levels. BMC Genomics. 2013, 14 (1): 185-10.1186/1471-2164-14-185.
Reichard M, Polacik M, Sedlacek O: Distribution, colour polymorphism and habitat use of the African killifish Nothobranchius furzeri, the vertebrate with the shortest life span. J Fish Biol. 2009, 74 (1): 198-212. 10.1111/j.1095-8649.2008.02129.x.
Bartakova V, Reichard M, Janko K, Pola Ik M, Bla Ek R, Reichwald K, Cellerino A, Bryja J: Strong population genetic structuring in an annual fish, Nothobranchius furzeri, suggests multiple savannah refugia in southern Mozambique. BMC Evol Biol. 2013, 13 (1): 196-10.1186/1471-2148-13-196.
Dorn A, Ng'oma E, Janko K, Reichwald K, Polacik M, Platzer M, Cellerino A, Reichard M: Phylogeny, genetic variability and colour polymorphism of an emerging animal model: the short-lived annual Nothobranchius fishes from southern Mozambique. Mol Phylogen Evol. 2011, 61 (3): 739-749. 10.1016/j.ympev.2011.06.010.
Valenzano DR, Kirschner J, Kamber RA, Zhang E, Weber D, Cellerino A, Englert C, Platzer M, Reichwald K, Brunet A: Mapping loci associated with tail color and sex determination in the short-lived fish Nothobranchius furzeri. Genetics. 2009, 183 (4): 1385-1395. 10.1534/genetics.109.108670.
Terzibasi E, Valenzano DR, Benedetti M, Roncaglia P, Cattaneo A, Domenici L, Cellerino A: Large differences in aging phenotype between strains of the short-lived annual fish Nothobranchius furzeri. PLoS One. 2008, 3 (12): e3866-10.1371/journal.pone.0003866.
Murisier F, Beermann F: Genetics of pigment cells: lessons from the tyrosinase gene family. Histol Histopathol. 2006, 21 (5): 567-578.
Baumgart M, Groth M, Priebe S, Savino A, Testa G, Dix A, Ripa R, Spallotta F, Gaetano C, Ori M, Eva Terzibasi T, Reinhard G, Matthias P, Alessandro C: RNA-seq of the aging brain in the short-lived fish N. furzeri - conserved pathways and novel genes associated with neurogenesis. Aging Cell. 2014, doi:10.1111/acel.12257. [Epub ahead of print]
Huang DW, Sherman BT, Lempicki RA: Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008, 4 (1): 44-57. 10.1038/nprot.2008.211.
del Marmol V, Beermann F: Tyrosinase and related proteins in mammalian pigmentation. FEBS Lett. 1996, 381 (3): 165-168. 10.1016/0014-5793(96)00109-3.
Cook AL, Smith AG, Smit DJ, Leonard JH, Sturm RA: Co-expression of SOX9 and SOX10 during melanocytic differentiation in vitro. Exp Cell Res. 2005, 308 (1): 222-235. 10.1016/j.yexcr.2005.04.019.
Murisier F, Guichard S, Beermann F: The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment Cell Res. 2007, 20 (3): 173-184. 10.1111/j.1600-0749.2007.00368.x.
Lamason RL: SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science (New York, NY). 2005, 310: 1782-1786. 10.1126/science.1116238.
Hammond CL, Hinits Y, Osborn DPS, Minchin JEN, Tettamanti G, Hughes SM: Signals and myogenic regulatory factors restrict Pax3 and Pax7 expression to dermomyotome-like tissue in zebrafish. Dev Biol. 2007, 302 (2): 504-521. 10.1016/j.ydbio.2006.10.009.
Minchin JE, Hughes SM: Sequential actions of Pax3 and Pax7 drive xanthophore development in zebrafish neural crest. Dev Biol. 2008, 317 (2): 508-522. 10.1016/j.ydbio.2008.02.058.
Medic S, Rizos H, Ziman M: Differential PAX3 functions in normal skin melanocytes and melanoma cells. Biochem Biophys Res Commun. 2011, 411 (4): 832-837. 10.1016/j.bbrc.2011.07.053.
Wu X, Bowers B, Rao K, Wei Q, Hammer JA: Visualization of melanosome dynamics within wild-type and dilute melanocytes suggests a paradigm for myosin V function In vivo. J Cell Biol. 1998, 143 (7): 1899-1918. 10.1083/jcb.143.7.1899.
Wu XS, Masedunskas A, Weigert R, Copeland NG, Jenkins NA, Hammer JA: Melanoregulin regulates a shedding mechanism that drives melanosome transfer from melanocytes to keratinocytes. Proc Natl Acad Sci U S A. 2012, 109 (31): E2101-E2109. 10.1073/pnas.1209397109.
Baumgart M, Groth M, Priebe S, Appelt J, Guthke R, Platzer M, Cellerino A: Age-dependent regulation of tumor-related microRNAs in the brain of the annual fish Nothobranchius furzeri. Mech Ageing Dev. 2012, 133 (5): 226-233. 10.1016/j.mad.2012.03.015.
Robinson MD, Oshlack A: A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11 (3): R25-10.1186/gb-2010-11-3-r25.
Anders S, Huber W: Differential expression analysis for sequence count data. Genome Biol. 2010, 11 (10): R106-10.1186/gb-2010-11-10-r106.
Benjamini Y, Hochberg Y: Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stati Soc B. 1995, 57 (1): 289-300.
MGFLI. count_genes_per_transcript.pl. https://github.com/MGFLI/RNAseq/tree/master
Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, Huang Da HC, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R: DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35 (suppl 2): W169-W175.
Koia JH, Moyle RL, Botella JR: Microarray analysis of gene expression profiles in ripening pineapple fruits. BMC Plant Biol. 2012, 12: 240-10.1186/1471-2229-12-240.
Pfaffl MW, Horgan GW, Dempfle L: Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30 (9): 1-10.
Acknowledgments
We thank Mario Baumgart for advice on RNA isolation and cDNA synthesis, Daniel Mertten for RNA isolation, Sabine Matz for expert technical assistance throughout the project, Bianca Lanick and Christin Hahn for help with fish husbandry.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AC conceived the project. All authors contributed to experimental design. EN bred the fish, prepared samples (tissue collection, RNA isolation and pooling), and performed GO analysis. MG conducted RNA-seq (library preparation, read mapping, DEG analysis and hierarchical clustering). RR carried out RT-qPCR validation. EN, AC, and MG interpreted results. MM contributed to project direction and critical review of manuscript. AC and EN wrote the draft manuscript. All authors revised the manuscript and approved the final version.
Electronic supplementary material
12864_2014_6450_MOESM1_ESM.csv
Additional file 1: CSV: List of 210 differentially expressed genes identified in the intersection of DESeq and edgeR.(CSV 27 KB)
12864_2014_6450_MOESM2_ESM.csv
Additional file 2: CSV: DAVID Functional Annotation Clustering results of up-regulated (A) and down-regulated (B) gene lists. Only gene groups containing significant GO terms are shown. The genes are listed below each cluster. (CSV 4 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Ng’oma, E., Groth, M., Ripa, R. et al. Transcriptome profiling of natural dichromatism in the annual fishes Nothobranchius furzeri and Nothobranchius kadleci. BMC Genomics 15, 754 (2014). https://doi.org/10.1186/1471-2164-15-754
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-754