
Abstract
Background
Myzus persicae, the green peach aphid, is a polyphagous herbivore that feeds from hundreds of species of mostly dicot crop plants. Like other phloem-feeding aphids, M. persicae rely on the endosymbiotic bacterium, Buchnera aphidicola (Buchnera Mp), for biosynthesis of essential amino acids and other nutrients that are not sufficiently abundant in their phloem sap diet. Tobacco-specialized M. persicae are typically red and somewhat distinct from other lineages of this species. To determine whether the endosymbiotic bacteria of M. persicae could play a role in tobacco adaptation, we sequenced the Buchnera Mp genomes from two tobacco-adapted and two non-tobacco M. persicae lineages.
Results
With a genome size of 643.5 kb and 579 predicted genes, Buchnera Mp is the largest Buchnera genome sequenced to date. No differences in gene content were found between the four sequenced Buchnera Mp strains. Compared to Buchnera APS from the well-studied pea aphid, Acyrthosiphon pisum, Buchnera Mp has 21 additional genes. These include genes encoding five enzymes required for biosynthesis of the modified nucleoside queosine, the heme pathway enzyme uroporphyrinogen III synthase, and asparaginase. Asparaginase, which is also encoded by the genome of the aphid host, may allow Buchnera Mp to synthesize essential amino acids from asparagine, a relatively abundant phloem amino acid.
Conclusions
Together our results indicate that the obligate intracellular symbiont Buchnera aphidicola does not contribute to the adaptation of Myzus persicae to feeding on tobacco.
Similar content being viewed by others

Background
Intraspecific variation in host plant use is common and well-documented in aphids [e.g. [1–5], and recently reviewed by [6]. One factor that has been demonstrated to greatly impact host plant use in the pea aphid, Acyrthosiphon pisum, is the presence of the secondary bacterial symbiont, Regiella insecticola[7–9]. In an elegant study that first eliminated R. insecticola from a naturally infected A. pisum lineage and then re-introduced infection of R. insecticola to the cured lineage, Tsuchida and colleagues demonstrated that the ability of A. pisum genotype TUt to utilize white clover (Trifolium repens) as a host is greatly facilitated by R. insecticola infection yet see [10]. Whereas secondary bacterial symbionts are commonly associated with A. pisum, they are less commonly found in other aphid species, including the cosmopolitan and polyphagous green peach aphid, Myzus persicae[11]. However, like A. pisum, M. persicae possesses the obligate intracellular bacterial symbiont Buchnera aphidicola, which has been implicated in intraspecific variation of the host nutritional requirements [12].
Almost all aphids rely on Buchnera for the biosynthesis of essential amino acids that are not sufficiently abundant in phloem [13–15]. Whole genome sequencing of Buchnera from multiple aphid species demonstrated clear interspecific variation in the ability of Buchnera to synthesize amino acids [16–20]. For example Buchnera Sg from the monocot-feeding aphid Schizaphis graminum has accumulated mutations in five of the genes required for the reduction of sulfur and cysteine biosynthesis [17], thus constraining S. graminum to a diet with sufficient sources of fixed sulfur [21]. Consistent with this, interspecific variation in aphid performance on artificial diet provides strong evidence that, in a range of aphid species, host plant adapted lineages vary in their amino acid biosynthetic abilities [e.g. [12, 22–24]. More recently, Vogel and Moran [12] identified six A. pisum lineages that varied in their dietary essential amino acid requirements. Using DNA sequencing and phenotyping of F1s from interclonal crosses they were able to demonstrate that, in the case of one of these six A. pisum lineages, dietary arginine requirements were determined by a mutation in the argC gene of its Buchnera endosymbiont.
M. persicae is a phloem-feeding insect that is found in all temperate regions of the world and infests hundreds of mostly dicot plant species, including many food and ornamental crops [25]. Although, as a species, M. persicae has a very broad host range, there are significant differences in host plant utilization and even specialization among M. persicae lineages [e.g. [1, 26–29]. The best studied host plant specialized lineages of M. persicae are those that thrive on tobacco [30–32]. Typically, M. persicae lineages found on tobacco are red and have relatively high nicotine tolerance [33], whereas non-tobacco lineages are green through yellow in color and have lower tolerance to nicotine [34]. Although tobacco-adapted lineages have been shown to be morphologically and genetically distinct from non-tobacco lineages of M. persicae, other genetic evidence clearly demonstrates maintenance of gene flow between tobacco-adapted and non-tobacco lineages [27, 35, 36].
Motivated by the possibility that differences in host plant specialization in M. persicae correlate with differences in the metabolic capabilities of the primary endosymbiont we present here the whole genome sequence of Buchnera aphidicola Mp (Buchnera Mp), the previously unsequenced obligate intracellular symbiont of M. persicae, from two tobacco-adapted aphid lineages and two aphid lineages collected from pepper and potato, respectively. These three plant species belong to different genera in the Solanaceae family, and are therefore distinct, but nevertheless closely related. We find that, at 643,504 bp the Buchnera Mp genome is the largest of all fully-sequenced Buchnera genomes. In addition to the large circular chromosome, the Buchnera Mp genome includes the leucine (pLeu) and tryptophan (pTrp) biosynthesis plasmids that appear to be common to all Buchnera from the Aphidini [37]. Although the Buchnera Mp genome differs from other sequenced Buchnera genomes in the presence or absence of key genes, we found only limited within-species differences in DNA sequence and no differences in gene content between Buchnera Mp lineages in aphids collected from solanaceous tobacco and non-tobacco hosts.
Results and discussion
Assembly of the four Buchnera Mp genomes and their associated plasmids
We generated Roche 454 and Illumina sequence data for all four Buchnera Mp lineages. Total read number for the Roche 454 libraries, with an average read length of 218 bp, ranged from 72,240 – 86,041 (Table 1). Using a de novo assembly approach, more than 67% of all Roche 454 reads for each Buchnera Mp strain were assembled into contigs (Table 1). For each genome, the contigs homologous to Buchnera APS (PRJNA57805) were identified by BLAST search, and syntenic contigs were assembled into Buchnera Mp genome scaffolds that covered 90% of the anticipated genome of ~640 kbp – a size estimate derived from the genomes of Buchnera from A. pisum (PRJNA57805) and Schizaphis graminum (PRJNA57913). Gap regions in the genome scaffolds were filled using an overlap–consensus approach with the 36 bp single–end Illumina libraries that had generated 3,801,794 – 10,435,517 reads (Table 1 and see Materials and Methods). This overlap-consensus approach to gap filling was applied to 15 gaps in the Buchnera Mp USDA chromosome, 137 gaps in the W106 chromosome, 12 gaps in the F009 chromosome and 146 gaps in the G002 chromosome. Thus the draft genome sequences for each lineage comprised the joint assembly of Roche 454 reads and Illumina reads. Following assembly of closed bacterial chromosomes, closed leucine plasmids, and open tryptophan plasmids for each of the four Buchnera Mp lineages, we searched our assembled data for any contigs that might represent novel Buchnera plasmids or secondary bacterial symbionts. We did not find any evidence of novel plasmids or secondary symbionts in any of the assemblies.
The quality of the draft genomes was measured by coverage of Roche 454 reads and Illumina reads. The average of Roche 454 reads on the assembled genome sequences was 20×, while the average Illumina coverage was 200×. Surprisingly, some regions that were shared across all four Buchnera Mp strains were characterized by low read coverage. All of these low read coverage regions were poly-A rich regions. Roche 454 sequencing technology is unreliable for sequencing AT rich regions and thus, because of the poor quality of the 454 data it was difficult to map the Illumina data to these AT rich genomic regions. Thus, for genomic regions that had less than 21× Illumina coverage, we removed the 454 data, making a gap in the assembly that we then filled using the overlap-consensus applied above. Following this, we used Sanger sequencing to verify any low-coverage genomic regions. After validation, a final genome sequence was constructed for each Buchnera Mp chromosome and plasmid. These sequences have been deposited in Genbank under the following accessions: For the tobacco strains - BUMPUSDA,CP002697; BUMPUSDA_pLeu CP002698; BUMPUSDA_pTrp JF928419; BUMPW106 CP002699; BUMPW106_pLeu CP002700; BUMPW106_pTrp JF928420; for the strain from pepper - BUMPG002 CP002701; BUMPG002_pLeu CP002702; BUMPG002_pTrp JF928418; and for the strain from potato - BUMPF009 CP002703; BUMPF009_pLeu CP002704; BUMPF009_pTrp JF928417.
All four Buchnera Mp genomes contain an identical gene set
This project was motivated by the possibility that differences in host plant specialization in M. persicae correlate with differences in the metabolic capabilities of the primary symbiont, Buchnera Mp. We found no differences in gene presence or absence across all four Buchnera Mp strains. Of the 582 protein coding genes in the tobacco adapted Buchnera Mp USDA genome, 312 are located on the positive strand, and 270 are on the negative strand. The relative positions of these genes are identical across all four Buchnera Mp strains. Differences in DNA sequence among the four strains are minimal (Table 2) with just under 13% of genes (74/581) containing nonsynonymous coding sequence differences in one or more of the four Buchnera Mp genomes (Figure 1, Table 3). We tested the distribution of nonsynonymous mutation on the genomes of Buchnera Mp strains W106, F009 and G002 by random statistical sampling [38] and cannot reject the hypothesis that nonsynonymous substitutions are randomly distributed (W106 p = 0.506, F009 p = 0.529 and G002 p = 0.522). Of the genes that contain nonsynonymous mutations, thirteen mutations affecting twelve genes are predicted by PROVEAN analysis [39] to have a deleterious effect on protein function (Additional file 1: Table S3). Of these thirteen genes, seven (clpP, dnaE, mrcB, rpoB, ybgI, yggJ and uppS) have functions that are internal to Buchnera, while the remaining five (cmk, dut, fabD, pta and thrC) are associated more directly with metabolic processes central to the symbiosis. Mutations in two genes are noteworthy. First, deleterious mutations in ybgI a cytoplasmic metal ion binding protein were identified in both non-tobacco adapted lineages (F009 and G002)(Additional file 1: Table S3 – marked by §). Second, the two non-tobacco adapted lineages share the same non-synonymous mutation in thrC (Additional file 1: Table S3 – marked by *). An amino acid biosynthesis gene, thrC is the final gene in the threonine biosynthesis pathway ([40] and see [41]). Threonine is one of the ten essential amino acids synthesized by Buchnera, suggesting that the non-tobacco lines of M. persicae examined here may have different threonine dietary requirements, an hypothesis that can be tested experimentally in future work.

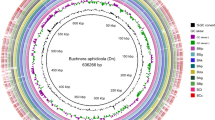
Maps of Buchnera Mp USDA, W106, F009, and G002 strains. The outside green wavy line is the AT frequency across the Buchnera Mp USDA genome. The three grey circles associated with the green wavy line mark 0, 0.5 and 1.0 AT frequency (from inside to outside). AT frequency was calculated using a 100 bp sliding-window with a window step of 10 bp. The red blocks on the two red circles are the annotated protein coding genes of the BuMp_USDA genome; the blue bars between the two red circles mark tRNAs and rRNAs. The three innermost orange circles represent the W106, F009 and G002 genomes with the orange bars on these circles indicating synonymous substitutions and the purple bars indicating nonsynonymous substitutions relative to BuMP_USDA.
Overall, comparison of the four Buchnera Mp genomes shows that the two tobacco-adapted strains (USDA and W106) are more closely related to one another than to F009 and G002 (Tables 3 and 2). This is consistent with the hypothesis that the ability to feed efficiently on tobacco arose relatively recently in M. persicae and has spread around the world in the past few decades [30, 42]. However, given that the overall gene content of the four Buchnera Mp strains is the same and only one gene central to the symbiosis shows a possible functional difference between tobacco and non-tobacco strains of Buchnera Mp, it is unlikely that changes within the Buchnera genome account for the ability of the aphids to feed from tobacco, thus any advantage Buchnera may provide to tobacco adapted lineages would need to result from differential gene expression. That said, previous analyses of global transcription in Buchnera demonstrate only a weak or limited capacity for transcriptional regulation of gene expression [43–48] and thus, we suggest that the ability of some M. persicae lineages to feed on tobacco results from differences in aphid genome architecture.
The Buchnera Mp genome, the largest Buchnera genome to date, retains metabolic capabilities lost in other Buchnera lineages
The Buchnera Mp genome shares features that have previously been described for other Buchnera species [16–18, 49–51], including a great reduction in genome size and number of genes relative to Escherichia coli, low GC content, and a strong bias toward the use of AT-rich codons in coding regions. With 643.5 kb and 579 predicted genes, Buchnera Mp is the largest sequenced Buchnera genome. Buchnera Mp retains more genes homologous to E. coli and has more protein coding genes than all other sequenced Buchnera genomes. Of particular interest is the fact that the gene profile of Buchnera Mp differs from those of other sequenced Buchnera strains. Of the 581 coding sequences of Buchnera Mp 96% (561) are orthologous to genes found in the second-largest Buchnera strain, Buchnera APS from A. pisum, an aphid that feeds on legumes. The remaining 4% correspond to 21 coding sequences (Additional file 1: Table S2, Figure 2). These 21 genes include two groups that are of particular biological interest: (1) a cluster of queuosine biosynthesis genes, and (2) two protein coding genes that are very rare among all sequenced Buchnera genomes. The queuosine biosynthesis cluster comprises five genes (equivalents of E. coli genes queC, ygcF(queE), queF, sscR, and folE, Figure 2) required for the biosynthesis of queuosine, a modified tRNA nucleoside found in bacteria and eukaryotes. Although absence of the five queuosine biosynthesis genes in Buchnera APS shows that they are not vital for Buchnera per se, genes homologs to all five of these genes are found in Buchnera Sg from S. graminum (AE013218.1), an aphid that feeds on monocots. Animals are unable to synthesize queuosine and must obtain it from their diets. However, given that it is not known whether queuosine can be transferred from Buchnera to its host, it is not clear whether queuosine biosynthetic capability impacts the symbiosis of Buchnera with their host aphids.

Map of global KEGG metabolic pathway present in Buchnera Mp showing genes present in Buchnera Mp but absent in Buchnera APS (salmon color), genes with nonsynonymous mutations among the four Buchnera Mp strains (blue color), genes with nonsynonymous mutations among the four Buchnera Mp strains that are also absent in Buchnera APS (orange color). Map generated from genome sequence of BuMP_USDA.
The two protein coding genes that are very rare among all sequenced Buchnera genomes are a predicted uroporphyrinogen III synthase (HemD, EC 4.2.1.75, USDA_CDS_00013) and a predicted asparaginase (ansA, EC 3.5.1.1, USDA_CDS_00060). Uroporphyrinogen III synthase is part of the incomplete heme biosynthetic pathway of Buchnera. There is no functional HemD encoded in the genome of Buchnera APS. However, BLASTp analysis shows the presence of pseudogenized HemD in Buchnera from A. pisum strains LSR1 (NZ_ACFK01000001.1), TLW03 (NC_017252.1) and JF99 (NC_017253.1), suggesting a relatively recent loss of this enzymatic function. The retention of an incomplete heme biosynthesis pathway across all sequenced Buchnera genomes shows that the host must supply metabolic precursors for these co-factors. Like other Buchnera, the Buchnera Mp genome encodes CysG, which catalyzes the multistep conversion of uroporphyrinogen III into siroheme, an essential cofactor for sulfite reductase and nitrite reductase. However, due to the presence of HemD (uroporphyrinogen III synthase) Buchnera Mp can receive metabolic precursors from its aphid host at an earlier step in the pathway. Since the Buchnera Mp genome encodes sulfite reductase, but not nitrite reductase, the main function of siroheme synthesis is likely the assimilation of inorganic sulfur, making the presence of siroheme essential for the production of cysteine and methionine [52].
ansA, which encodes an asparaginase, is only known from one other Buchnera genome, that of Acyrthosiphon kondoi (NC_017256.1). As one of the most abundant phloem transport amino acids in plants [53], asparagine should be utilized efficiently in the aphid-Buchnera system. Current models for amino acid biosynthesis in A. pisum indicate that aphid asparaginase converts asparagine to ammonia and aspartate, which is transported into Buchnera APS for the biosynthesis of threonine and lysine [52, 54, 55]. However, since Buchnera does not encode asparagine biosynthesis enzymes, there must be at least some transport of this amino acid from the aphid to the endosymbionts. The presence of ansA in the Buchnera Mp genome indicates that phloem-derived asparagine may be transported directly to Buchnera Mp for the biosynthesis of other amino acids. Similar to the three predicted A. pisum asparaginases (XP_001944435, XP_001949831, and XP_001942778), three M. persicae ESTs (EE262575, ES22392, and ES223192) arise from genes that encode predicted asparaginases. Therefore, this enzymatic function is not missing from M. persicae, despite the fact that it also is encoded by the endosymbiont.
To date, the only significant transcriptional regulation of Buchnera amino acid biosynthesis was observed for the methionine biosynthesis pathway of Buchnera Sg [45]. This regulatory activity was ascribed to the metR, one of the few bacterial transcriptional regulators retained in the Buchnera genome. However, although metR is present in Buchnera Sg and Buchnera Mp, it is not found in the other Buchnera genomes (Additional file 1: Table S2). S. graminum and M. persicae have wider host ranges than A. pisum, Cinara cedri, and Baizongia pistaceae. As such, they would be exposed to a broader range of plant sulfur-transport metabolites and might have a greater need to regulate the biosynthesis of methionine. For instance, glutathione and S-methylmethionine are the two most abundant forms of reduced sulfur in plant phloem [56]. However, whereas S-methylmethionine can likely be converted to methionine by predicted aphid homocysteine methyltransferases, breakdown of glutathione releases cysteine, which would need to be converted to methionine by the Buchnera endosymbionts. Therefore, biosynthesis of methionine in Buchnera would need to be regulated based on the relative abundance of glutathione and S-methylmethionine in different host plants.
The Buchnera Mp genome contains a leucine and a tryptophan biosynthesis plasmid
The plasmid pLeu was assembled and annotated completely for the four Buchnera Mp strains, and the gene profile is similar to that of Buchnera APS (Table 4). The plasmid pTrp, which in Buchnera characteristically contains a repeat of the tryptophan operon [16], was partially assembled in the four Buchnera Mp strains. The gene content and gene order of pTrp matches that of Buchnera APS. Therefore these two plasmids seem to be fulfilling their roles in the Buchnera Mp strains without further variation and evolution.
Conclusion
Our sequencing of the Buchnera endosymbiont genomes from four strains of Myzus persicae shows 100% conservation of gene content. This lack of differences between tobacco-adapted and non-adapted strains of Buchnera Mp suggests that the endosymbionts play no role in the ability of some M. persicae lineages to grow well on tobacco. The presence of metR and ansA in the genome of Buchnera Mp, but not in the genome of Buchnera APS, may allow more adaptive use of dietary amino acids and contribute to the ability of M. persicae grow on a wider variety of host plants than the legume specialist A. pisum.
Materials and methods
Selection of aphid lineages
We sequenced the Buchnera genome from two isofemale lineages of M. persicae collected from tobacco (USDA [from greenhouse tobacco in Ithaca, NY, 2003, 57]) and W106 (from field-grown tobacco in Windsor, CT, 2007)) and two M. persicae isofemale lineages from non-tobacco hosts (G002 (from field-grown pepper in Geneva, NY, 2003), and F009 (from field-grown potato in Freeville, NY, 2003; both from [57]). Since their collection, aphid lineages have been maintained in the laboratory on cabbage seedlings (Brassica oleracea var. Wisconsin Golden Acre) at 20°C under long day conditions (16 hours light: 8 hours dark). To ensure the maintenance of clonal lineages, microsatellite genotyping is performed biannually (Additional file 1: Table S1).
Microsatellite genotyping of aphid lineages
Aphid cultures in the lab of Alex Wilson are genotyped biannually to ensure maintenance of clonal integrity. Myzus persicae cultures are genotyped at six loci: M40 (forward primer labeled with VIC, M40-for-VIC) and M63 (M63-for-6-FAM) from [58]; myz2 (reverse primer labeled with PET, myz2–rev-PET), myz9 (myz9-for-VIC), myz25 (myz25-for-NED), and S17b (S17b-for-NED) from [59]. All six loci are amplified in a single multiplexed PCR reaction containing 1 × Qiagen PCR Buffer, 2 mM MgCl2, 0.21 mM of each dNTP, 1 unit of Qiagen Taq DNA polymerase, 0.04 μM of each M40f/r, myz9f/r, myz25f/r, and S17bf/r, 0.08 μM of each myz2f/r, 0.17 μM of each M63f/r and 10 ng of genomic DNA to a final volume of 12 μL. The following touchdown PCR program is run: 94°C for 2 min, followed by 8 cycles of 94°C for 30 sec, touch-down annealing for 1 min at 62°C stepping down 1°C per cycle for 8 cycles, 72°C for 45 sec, followed by 22 cycles of 94°C for 30 sec, 55°C for 1 min, 72°C for 45 sec with a final extension of 72°C for 6 min. PCR products are diluted 1:50 prior to analysis on an Applied Biosystems 3130xl Genetic Analyzer using the Applied Biosystems GeneScan 500 LIZ Size Standard.
Isolation of Buchnera DNA
Buchnera are obligately symbiotic bacteria that have never successfully been cultured. Thus, prior to DNA extraction aphid material was subject to enrichment for intact Buchnera. From each lineage, 1.5-2.5 g of aphids was crushed in a cold mortar in 20 ml Buffer A (25 mM KCl, 35 mM Tris–HCl, 100 mM EDTA, 250 mM sucrose, pH 8.0). The homogenate was passed twice through a 100 μm pore-sized nylon filter (Millipore) and centrifuged at 1,500 g at 4°C for 10 min. The pellet was resuspended in 25 ml Buffer A and passed through decreasing pore-sized filters: once through a 20 μm nylon filter (Millipore), twice through an 11 μm nylon filter (Millipore), twice through an 8 μm polycarbonate filter (Whatman, Schleicher & Schuell), three times through a 5 μm polycarbonate Isopore filter (Millipore) and three times through a 3 μm polycarbonate filter (Whatman, Schleicher and Schuell). The filtrate was centrifuged at 1,500 g at 4°C for 25 min and the pellet resuspended in 3 ml Buffer A, and further purified using a Percoll gradient (5% PEG6000, 1% BSA, 1% Ficoll, 250 mM sucrose with 27 or 70% Percoll) by centrifugation at 12,000 g at 4°C for 15 min. Buchnera cells were recovered from between the 27 and 70% Percoll layers, diluted in 10 ml Buffer A and centrifuged at 1,500 g at 4°C for 15 min. The cell pellet was washed twice with 2 ml Buffer A. Following washing DNA was extracted from the cell pellet using a Qiagen DNeasy column with inclusion of the RNase A treatment and eluted twice into 100 μl Qiagen Buffer AE yielding an average of 12 μg of Buchnera DNA per gram of aphid fresh weight.
To verify our extraction protocol was sufficiently enriching for Buchnera DNA relative to aphid DNA, we utilized real time quantitative PCR (RT-qPCR) to measure the abundance of Buchnera single copy gene dnaK (using primers dnaK2F 5′-GATTGTCTTCGGCTGTTG-3′ and dnaK2R 5′- GTCACTCCTTTATCACTTGG-3′) relative to the abundance of aphid single copy gene elongation factor 1α (using primers AWRT002F 5′- CTGATTGTGCTGTGCTTATTG-3′ and AWRT002R 5′- CAAGGTGAAAGCCAATAGAGC-3′). We found that we can reliably increase Buchnera copy number in our enriched sample relative to DNA extracted from whole aphids by 100 fold. Meaning that in any given sample we conservatively estimated that Buchnera DNA would represent 15% of the reads. An estimate we then used to determine the amount of sequencing that would be required to generate high quality genomes for each of the four Buchnera Mp lines.
Next-generation genome sequencing of Buchnera aphidicola Mp
For each Buchnera strain we generated 118 to 124 Mb of DNA sequence data using Roche 454 LR70 sequencing performed by the Environmental Genomics Core Facility at the University of South Carolina, and 136 to 375 Mb of DNA sequence data using an Illumina Genome Analyzer at the Cornell University Life Sciences Core Laboratories Center. DNA for Roche 454 sequencing was prepared for sequencing following the manufacture’s instructions. DNA for Illumina sequencing was randomly sheared by nebulization prior to library preparation using an Illumina Genomic DNA sample preparation kit according to the manufacture’s protocol.
Verification of genome sequence and assembly by Sanger sequencing
Target fragments in regions with less than 21× Illumina coverage were amplified using the following touchdown PCR program: 94°C for 2 min, followed by 8 cycles of 94°C for 30 sec, touch-down annealing for 1 min at 62°C stepping down 1°C per cycle for 8 cycles, 72°C for 45 sec, followed by 31 cycles of 94°C for 30 sec, 55°C for 1 min, 72°C for 45 sec with a final extension of 72°C for 6 min. PCR amplification was performed in 10 μL reactions containing 1 × Qiagen PCR Buffer, 0.25 mM of each dNTP, 0.5 μM of each primer, 0.2 units of Qiagen Taq DNA Polymerase, and 10 ng of genomic DNA. PCR products were purified in 7 μL reactions using 0.1 units of Exonuclease I and 0.1 units of Shrimp Alkaline Phosphatase at 37°C for 15 min followed by enzyme deactivation at 80°C for 15 min. Cycle sequencing using the Applied Biosystems BigDye® Terminator v3.1 Cycle Sequencing Kit was performed in 10 μL reactions with 2 μL of purified PCR product, 0.375 μL BigDye® Terminator v3.1, 1× Sequencing Buffer, and 1 μM of primer under the following thermal cycling conditions: 35 cycles of 94°C for 30 sec, 50°C for 15 sec, and 60°C for 4 min. DNA sequencing reactions were purified using Sephadex G-50 Fine DNA Grade, dehydrated and resuspended in 10 μL Applied Biosystems Hi-Di™ Formamide prior to analysis on the Applied Biosystems 3130xl Genetic Analyzer.
Genome assembly
Roche 454 reads were assembled using Roche Genome Sequencer FLX software. Facilitated by the fact that genome evolution in Buchnera is characterized by conservation of gene order [19], we built a genome scaffold for each Buchnera Mp strain by BLASTing assembled 454 contigs against the Buchnera APS genome (NC_002528). Each scaffold contained gaps. Gap size was inferred by the distance between contigs assembled against the Buchnera APS genome. Illumina reads were assembled across gaps using the overlap-consensus approach [60]. Briefly, a 30 bp fragment with a high sequencing quality score from the 3′ end of each contig upstream of a gap was selected as the assembly seed. All Illumina reads and their complementary sequences were maintained in a repository for Illumina read assembly. The repository was searched for Illumina reads containing a 30 bp fragment matching the seed, and up to 2 mismatches were allowed in the matching process. A base after the matched 30 bp fragment of each Illumina read was recorded, and the consensus nucleotide was determined by the majority rule on the basis of recorded bases of all matched reads. The matched reads (and corresponding complementary sequences) from which the last base was recorded were removed from the repository, and not used for further assembly. The assembled consensus sequence was extended by adding the determined consensus nucleotide to 3′ end, and the seed was shifted a base to the 3′ end on the extended sequence after the addition of a base. This process was repeated until the consensus sequence reached the 5′ end of the other contig downstream of the gap.
We used MAQ [61] to map Illumina reads on the assembled sequences with up to 3 mismatches. The coverage of Illumina reads on each site was calculated and used to identify potential sequencing errors. Regions with low coverage of Illumina reads (<21×) were first screened by an in-house PERL script for sequencing errors, such as an indel or a wrong base call originated from Roche 454 reads. Errors attributed to the 454 sequence were corrected using the consensus sequence of Illumina reads. In regions where the errors couldn’t be corrected by the script due to the insufficient Illumina coverage, a Sanger PCR sequencing approach was applied.
Genome annotation
Gene models in the assembled genomes and plasmid scaffolds were predicted with FgenesB (http://www.softberry.com, utilizing the annotated genome of Buchnera aphidicola Bp (NC_004545) as a training set), GeneMark [62], and GLIMMER [63]. Predictions shared by all three gene prediction pipelines were considered candidate genes and annotated by running BLAST against the NCBI non-redundant protein databases (nr) and Buchnera aphidicola str. APS (taxid: 107806) proteins. All candidate genes with e-values less than 4e-04 to known proteins were considered Buchnera Mp coding sequences and annotated accordingly. Transfer RNAs (tRNAs) were predicted using the online version of tRNAscan [64], whereas rRNAs were annotated by BLAST search.
Manual curation
All genes that differed in length from the Buchnera APS models by >10%, together with any gene models that were interrupted by premature stop codons, were manually curated. These included 38 gene models across the four Buchnera Mp genomes. Assessment of automated gene model predictions during manual curation involved alignment of Buchnera Mp models against each other, as well as assessment of the similarity, nucleotide and amino acid percent identity and model length against those of Buchnera APS and the top E. coli hits. In some instances, it was necessary to perform Pfam domain searches [65] to confirm homology. Frequent manual curation issues included: Buchnera Mp models that were longer than Buchnera APS models, but close in length to the E. coli gene; these Buchnera Mp models were left unchanged. Buchnera Mp models that were longer than both Buchnera APS and E. coli homologs; these models were also left unchanged. Buchnera Mp models that were shorter than the Buchnera APS homolog; these were frequently extended to an upstream alternate start codon (often GTG and TTG) and occasionally the rare ATT start codon. Most commonly, manual curation involved indels in polyA tracts resulting in automated annotation either truncating the gene or splitting it into two separate partial gene models. In all cases, DNA sequence on either side of the polyA tract was conserved, and every indication was that the gene had been subject to purifying selection, remaining functional. In these cases, the length of the polyA tract was left unchanged but the gene models were merged or extended, resulting in a DNA sequence that codes for a gene in two reading frames. This feature of symbiotic bacterial genomes, including the genome of Buchnera aphidicola from Schizaphis graminum has been reported previously [66].
Pathway analysis
The functional annotation of the verified Buchnera Mp genes was performed using the KEGG Annotation Database (http://www.genome.jp/tools/kaas). We compared the query genes against the manually curated KEGG genes database and assigned KO (KEGG Orthology) to query genes. The information of pathways and associated KO were retrieved from KEGG web sites, and the candidate genes were grouped into KEGG pathways accordingly. The number of pathways, as well as the number of genes in each pathway, was counted for each strain. The pathway statistics of the four Buchnera Mp strains were compared with the other Buchnera strains and E. coli, to investigate the evolutionary patterns of pathways in our strains.
Analysis of non-synonymous mutations
The impact of non-synonymous mutations on proteins was predicted by PROVEAN [39]. The distributions of non-synonymous mutations across the W106, G002 and F009 genomes relative to USDA were tested for randomness [38]. The randomness of nonsynonymous mutations was evaluated by the runs.test function [67] in R [68].
Authors’ information
ZJ, SK and NT are computational scientists interested in genome assembly using next generation sequencing data and gene prediction. GJ studies insect/host plant interactions with a particular focus on insect elicitors of host defensive responses. DHJ worked as a technician in Wilson’s lab. TW was a graduate student research assistant in Wilson’s lab. Utilizing a diversity of experimental approaches from stable isotopes through whole genome sequencing, ACCW studies the metabolic collaboration of host/symbiont systems with a focus on nitrogen metabolism.
References
Blackman RL: Morphological discrimination of a tobacco-feeding form from Myzus persicae (Sulzer) (Hemiptera: Aphididae), and a key to New World Myzus (Nectarosiphon)species. Bull ent Res. 1987, 77: 713-730. 10.1017/S0007485300012219.
Sunnucks P, De Barro PJ, Lushai G, Maclean N, Hales DF: Genetic structure of an aphid studied using microsatellites: cyclic parthenogenesis, differentiated lineages, and host specialization. Mol Ecol. 1997, 6: 1059-1073. 10.1046/j.1365-294X.1997.00280.x.
Peccoud J, Figueroa CC, Silva AX, Ramirez CC, Mieuzet L, Bonhomme J, Stoeckel S, Plantegenest M, Simon JC: Host range expansion of an introduced insect pest through multiple colonizations of specialized clones. Mol Ecol. 2008, 17 (21): 4608-4618. 10.1111/j.1365-294X.2008.03949.x.
Valenzuela I, Ridland PM, Weeks AR, Hoffmann AA: Patterns of genetic variation and host adaptation in an invasive population of Rhopalosiphum padi (Hemiptera: Aphididae). Ann Entomol Soc Am. 2010, 103 (6): 886-897. 10.1603/AN09113.
Weng YQ, Perumal A, Burd JD, Rudd JC: Biotypic diversity in greenbug (Hemiptera: Aphididae): microsatellite-based regional divergence and host-adapted differentiation. J Econ Entomol. 2010, 103 (4): 1454-1463. 10.1603/EC09291.
Peccoud J, Simon JC, von Dohlen C, Coeur d'Acier A, Plantegenest M, Vanlerberghe-Masutti F, Jousselin E: Evolutionary history of aphid-plant associations and their role in aphid diversification. C R Biol. 2010, 333 (6–7): 474-487.
Tsuchida T, Koga R, Fukatsu T: Host plant specialization governed by facultative symbiont. Science. 1989, 2004: 303-
Ferrari J, Scarborough CL, Godfray HCJ: Genetic variation in the effect of a facultative symbiont on host-plant use by pea aphids. Oecologia. 2007, 153 (2): 323-329. 10.1007/s00442-007-0730-2.
Tsuchida T, Koga R, Matsumoto S, Fukatsu T: Interspecific symbiont transfection confers a novel ecological trait to the recipient insect. Biol Lett. 2011, 7 (2): 245-248. 10.1098/rsbl.2010.0699.
McLean AHC, van Asch M, Ferrari J, Godfray HCJ: Effects of bacterial secondary symbionts on host plant use in pea aphids. Proc R Soc B-Biol Sci. 2011, 278 (1706): 760-766. 10.1098/rspb.2010.1654.
von Burg S, Ferrari J, Muller CB, Vorburger C: Genetic variation and covariation of susceptibility to parasitoids in the aphid Myzus persicae: no evidence for trade-offs. Proc R Soc B-Biol Sci. 2008, 275 (1638): 1089-1094. 10.1098/rspb.2008.0018.
Vogel KJ, Moran NA: Sources of variation in dietary requirements in an obligate nutritional symbiosis. Proc R Soc B-Biol Sci. 2011, 278 (1702): 115-121. 10.1098/rspb.2010.1304.
Buchner P: Endosymbiosis of animals with plant microorganisms. 1965, New York: Interscience
Baumann P, Baumann L, Lai C-Y, Rouhbakhsh D, Moran NA, Clark MA: Genetics, physiology, and evolutionary relationships of the genus Buchnera: Intracellular symbionts of aphids. Annu Rev Microbiol. 1995, 49: 55-94. 10.1146/annurev.mi.49.100195.000415.
Douglas AE: Phloem-sap feeding by animals: problems and solutions. J Exp Bot. 2006, 57 (4): 747-754. 10.1093/jxb/erj067.
Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H: Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000, 407 (6800): 81-86. 10.1038/35024074.
Tamas I, Klasson L, Canback B, Naslund AK, Eriksson A-S, Wernegreen JJ, Sandstrom JP, Moran NA, Andersson SGE: 50 Million years of genomic stasis in endosymbiotic bacteria. Science. 2002, 296 (5577): 2376-2379. 10.1126/science.1071278.
van Ham RCHJ, Kamerbeek J, Palacios C, Rausell C, Abascal F, Bastolla U, Fernandez JM, Jimenez L, Postigo M, Silva FJ, et al: Reductive genome evolution in Buchnera aphidicola. Proc Natl Acad Sci U S A. 2003, 100 (2): 581-586. 10.1073/pnas.0235981100.
Pérez-Brocal V, Gil R, Ramos S, Lamelas A, Postigo M, Michelena JM, Silva FJ, Moya A, Latorre A: A small microbial genome: the end of a long symbiotic relationship?. Science. 2006, 314: 312-313. 10.1126/science.1130441. 13 October 2006
Lamelas A, Jose Gosalbes M, Moya A, Latorre A: New clues about the evolutionary history of metabolic losses in bacterial endosymbionts, provided by the genome of Buchnera aphidicola from the aphid Cinara tujafilina. Appl Environ Microbiol. 2011, 77 (13): 4446-4454. 10.1128/AEM.00141-11.
Moran NA, Degnan PH: Functional genomics of Buchnera and the ecology of aphid hosts. Mol Ecol. 2006, 15: 1251-1261.
Srivastava PN, Gao Y, Levesque J, Auclair JL: Differences in amino-acid requirements between 2 biotypes of the pea aphid. Acyrthosiphon pisum. Can J of Zool. 1985, 63 (3): 603-606. 10.1139/z85-087.
Wilkinson TL, Douglas AE: Phloem amino acids and the host plant range of the polyphagous aphid. Aphis fabae. Entomologia Exp et Appl. 2003, 106 (2): 103-113. 10.1046/j.1570-7458.2003.00014.x.
MacDonald SJ, Thomas GH, Douglas AE: Genetic and metabolic determinants of nutritional phenotype in an insect-bacterial symbiosis. Mol Ecol. 2011, 20 (10): 2073-2084. 10.1111/j.1365-294X.2011.05031.x.
Blackman RL, Eastop VF: Aphids on the world's crops: An identification and information guide. 2000, Chichester: John Wiley & Sons Ltd., 2
Margaritopoulos JT, Tsitsipis JA, Zintzaras E, Blackman RL: Host-correlated morphological variation of Myzus persicae (Hemiptera : Aphididae) populations in Greece. Bull Entomol Res. 2000, 90 (3): 233-244.
Margaritopoulos JT, Blackman RL, Tsitsipis JA, Sannino L: Co-existence of different host-adapted forms of the Myzus persicae group (Hemiptera: Aphididae) in southern Italy. Bull Entomol Res. 2003, 93: 131-135.
Margaritopoulos JT, Tsourapas C, Tzortzi M, Kanavaki OM, Tsitsipis JA: Host selection by winged colonisers within the Myzus persicae group: a contribution towards understanding host specialisation. Ecological Entomol. 2005, 30 (4): 406-418. 10.1111/j.0307-6946.2005.00700.x.
Vargas RR, Troncoso AJ, Tapia DH, Olivares-Donoso R, Niemeyer HM: Behavioural differences during host selection between alate virginoparae of generalist and tobacco-specialist Myzus persicae. Entomol Exp Appl. 2005, 116 (1): 43-53. 10.1111/j.1570-7458.2005.00311.x.
McPherson RM: Seasonal abundance of red and green morphs of the tobacco aphid (Homoptera, Aphididae) on flue-cured tobacco in Georgia. J Entomol Sci. 1989, 24 (4): 531-538.
Zepeda-Paulo FA, Simon J-C, Ramírez CC, Fuentes-Contreras E, Margaritopoulos JT, Sorenson CE, Wilson ACC, Briones LM, de Acevedo R, Ohashi DV, et al: The invasion route for an insect pest species: the tobacco aphid in the new world. Mol Ecol. 2010, 19 (21): 4738-4752. 10.1111/j.1365-294X.2010.04857.x.
Lai RQ, You MS, Lotz LAP, Vasseur L: Response of green peach aphids and other arthropods to garlic intercropped with tobacco. Agron J. 2011, 103 (3): 856-863. 10.2134/agronj2010.0404.
Nauen R, Strobel J, Tietjen K, Otsu Y, Erdelen C, Elbert A: Aphicidal activity of imidacloprid against a tobacco feeding strain of Myzus persicae (Homoptera: Aphididae) from Japan closely related to Myzus nicotianae and highly resistant to carbamates and organophosphates. Bull Entomol Res. 1996, 86 (2): 165-171. 10.1017/S0007485300052408.
Devine GJ, Harling ZK, Scarr AW, Devonshire AL: Lethal and sublethal effects of imidacloprid on nicotine-tolerant Myzus nicotianae and Myzus persicae. Pestic Sci. 1996, 48 (1): 57-62. 10.1002/(SICI)1096-9063(199609)48:1<57::AID-PS435>3.0.CO;2-9.
Margaritopoulos JT, Shigehara T, Takada H, Blackman RL: Host-related morphological variation within Myzus persicae group (Homoptera : Aphididae) from Japan. Appl Entomol Zool. 2007, 42 (2): 329-335. 10.1303/aez.2007.329.
Eastop VF, Blackman RL: Some new synonyms in Aphididae (Hemiptera : Sternorrhyncha). Zootaxa. 2005, 1089: 1-36.
Gil R, Sabater-Munoz B, Perez-Brocal V, Silva FJ, Latorre A: Plasmids in the aphid endosymbiont Buchnera aphidicola with the smallest genomes. A puzzling evolutionary story. Gene. 2006, 370: 17-25.
Bradley JV: Distribution-free statistical tests. 1968, New Jersey, USA: Prentice Hall
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP: Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012, 7 (10): e46688-10.1371/journal.pone.0046688.
Shigenobu S, Wilson ACC: Genomic revelations of a mutualism: the pea aphid and its obligate bacterial symbiont. Cell Mol Life Sci. 2011, 68: 1297-1309. 10.1007/s00018-011-0645-2.
Russell CW, Bouvaine S, Newell PD, Douglas AE: Shared metabolic pathways in a coevolved insect-bacterial symbiosis. Appl Environ Microbiol. 2013, 79: 6117-6123. 10.1128/AEM.01543-13.
Lampert EP, Dennis CA: Life history of two color morphs of the green peach aphid (Homoptera: Aphididae) on flue-cured tobacco. Tob Sci. 1987, 31: 91-93.
Wilson ACC, Dunbar HE, Davis GK, Hunter WB, Stern DL, Moran NA: A dual-genome microarray for the pea aphid, Acyrthosiphon pisum, and its obligate symbiont. Buchnera aphidicola. BMC Genomics. 2006, 7: 50-10.1186/1471-2164-7-50.
Wilcox JL, Dunbar HE, Wolfinger RD, Moran NA: Consequences of reductive evolution for gene expression in an obligate endosymbiont. Mol Microbiol. 2003, 48 (6): 1491-1500. 10.1046/j.1365-2958.2003.03522.x.
Moran NA, Dunbar HE, Wilcox JL: Regulation of transcription in a reduced bacterial genome: nutrient-provisioning genes of the obligate symbiont Buchnera aphidicola. J Bacteriol. 2005, 187 (12): 4229-4237. 10.1128/JB.187.12.4229-4237.2005.
Reymond N, Calevro F, Vinuelas J, Morin N, Rahbe Y, Febvay G, Laugier C, Douglas A, Fayard JM, Charles H: Different levels of transcriptional regulation due to trophic constraints in the reduced genome of Buchnera aphidicola APS. Appl Environ Microbiol. 2006, 72 (12): 7760-7766. 10.1128/AEM.01118-06.
Bermingham J, Rabatel A, Calevro F, Vinuelas J, Febvay G, Charles H, Douglas A, Wilkinson T: Impact of host developmental age on the transcriptome of the symbiotic bacterium Buchnera aphidicola in the pea aphid (Acyrthosiphon pisum). Appl Environ Microbiol. 2009, 75 (22): 7294-7297. 10.1128/AEM.01472-09.
Vinuelas J, Calevro F, Remond D, Bernillon J, Rahbe Y, Febvay G, Fayard JM, Charles H: Conservation of the links between gene transcription and chromosomal organization in the highly reduced genome of Buchnera aphidicola. BMC Genomics. 2007, 8: 143-10.1186/1471-2164-8-143.
Gil R, Sabater-Muñoz B, Latorre A, Silva FJ, Moya A: Extreme genome reduction in buchnera spp.: toward the minimal genome needed for symbiotic life. Proc Natl Acad Sci U S A. 2002, 99 (7): 4454-4458. 10.1073/pnas.062067299.
Moran NA, McLaughlin HJ, Sorek R: The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science. 2009, 323: 379-382. 10.1126/science.1167140.
Gosalbes MJ, Lamelas A, Moya A, Latorre A: The striking case of tryptophan provision in the cedar aphid Cinara cedri. J Bacteriol. 2008, 190 (17): 6026-6029. 10.1128/JB.00525-08.
Wilson ACC, Ashton PD, Calevro F, Charles H, Colella S, Febvay G, Jander G, Kushlan PF, Macdonald SJ, Schwartz JF, et al: Genomic insight into the amino acid relations of the pea aphid Acyrthosiphon pisum with its symbiotic bacterium Buchnera aphidicola. Insect Mol Biol. 2010, 19 (Suppl. 2): 249-258.
Coruzzi GM, Last RL: Amino acids. Biochemistry and Molecular Biology of Plants. Edited by: Buchanan RB, Gruissem W, Jones R. 2000, Rockville, MD: American Society of Plant Physiology Press, 358-410.
Hansen AK, Moran NA: Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc Natl Acad Sci USA. 2011, 108 (7): 2849-2854. 10.1073/pnas.1013465108.
Poliakov A, Russell CW, Ponnala L, Hoops HJ, Sun Q, Douglas AE, van Wijk KJ: Large-scale label-free quantitative proteomics of the pea aphid-Buchnera symbiosis. Mol Cell Proteomics. 2011, 10: 6-10.1074/mcp.M1110.007039–007033
Bourgis F, Roje S, Nuccio ML, Fisher DB, Tarczynski MC, Li CJ, Herschbach C, Rennenberg H, Pimenta MJ, Shen TL, et al: S-methylmethionine plays a major role in phloem sulfur transport and is synthesized by a novel type of methyltransferase. Plant Cell. 1999, 11 (8): 1485-1497.
Ramsey JS, Wilson ACC, de Vos M, Sun Q, Tamborindeguy C, Winfield A, Malloch G, Smith DM, Fenton B, Gray SM, et al: Genomic resources for Myzus persicae: EST sequencing, SNP identification, and microarray design. BMC Genomics. 2007, 8: 423-10.1186/1471-2164-8-423.
Sloane MA, Sunnucks P, Wilson ACC, Hales DF: Microsatellite isolation, linkage group identification and determination of recombination frequency in the peach-potato aphid, Myzus persicae (Sulzer) (Hemiptera: Aphididae). Genet Res, Camb. 2001, 77: 251-260.
Wilson ACC, Massonnet B, Simon J-C, Prunier-Leterme N, Dolatti L, Llewellyn KS, Figueroa CC, Ramirez CC, Blackman RL, Estoup A, et al: Cross-species amplification of microsatellite loci in aphids: assessment and application. Mol Ecol Notes. 2004, 4 (1): 104-109. 10.1046/j.1471-8286.2004.00584.x.
Myers EW: Toward simplifying and accurately formulating fragment assembly. J Comput Biol. 1995, 2: 275-290. 10.1089/cmb.1995.2.275.
Li H, Ruan J, Durbin R: Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18 (11): 1851-1858. 10.1101/gr.078212.108.
Lukashin AV, Borodovsky M: GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998, 26 (4): 1107-1115. 10.1093/nar/26.4.1107.
Delcher AL, Harmon D, Kasif S, White O, Salzberg SL: Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27 (23): 4636-4641. 10.1093/nar/27.23.4636.
Schattner P, Brooks AN, Lowe TM: The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33 (suppl 2): W686-W689.
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunesekaran P, Ceric G, Forslund K, et al: The Pfam protein families database. Nucleic Acids Res. 2010, 38: D211-222. 10.1093/nar/gkp985.
Tamas I, Wernegreen JJ, Nystedt B, Kauppinen SN, Darby AC, Gomez-Valero L, Lundin D, Poole AM, Andersson SGE: Endosymbiont gene functions impaired and rescued by polymerase infidelity at poly(A) tracts. Proc Natl Acad Sci USA. 2008, 105 (39): 14934-14939. 10.1073/pnas.0806554105.
Trapletti A, Hornik K: tseries: time series analysis and computational. 2013, finance: R package, 0.10-32 edn
Team RC: R: A language and environment for statistical computing. Edited by: Computing RFfS. 2013, Vienna, Austria
Acknowledgements
This project was supported by United States Department of Agriculture - National Institute of Food and Agriculture grants 2005-35604-15446 and 2010-65105-20558 to ACCW and GJ, by University of Miami Start-up Funds, a University of Miami General Research Support Award in the Natural Sciences and Engineering to ACCW, and the Paul and Maxine Frohring Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ACCW & GJ conceived of and designed the study. ZJ assembled and annotated the genomes and performed bioinformatic analyses in collaboration with SK, NT and ACCW. DHJ performed gap-closing Sanger sequencing and executed genome quality control analysis in collaboration with ACCW. TW isolated and prepared Buchnera DNA for genome sequencing in collaboration with ACCW. ACCW & ZJ drafted the manuscript. ACCW, SK, NT and GJ contributed to preparation of the final manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2013_5628_MOESM1_ESM.docx
Additional file 1: Table S1. Collection information and microsatellite genotype of Myzus persicae lineages hosting the four sequenced Buchnera Mp strains. Table S2. Twenty-one Buchnera Mp coding sequences whose counterparts are absent in Buchnera APS. Table S3. Thirteen nonsynonymous mutations in the Buchnera Mp genomes of lineages W106, F009 and G002 that are predicted to be deleterious. Figure S1. Nucleotide composition of all protein coding sequences in the genomes of Buchnera Mp USDA (BTI), Buchnera APS, Buchnera Cc and E. coli. Data shown by codon position and by all positions. (DOCX 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jiang, Z., Jones, D.H., Khuri, S. et al. Comparative analysis of genome sequences from four strains of the Buchnera aphidicola Mp endosymbion of the green peach aphid, Myzus persicae. BMC Genomics 14, 917 (2013). https://doi.org/10.1186/1471-2164-14-917
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-14-917