
Abstract
Enterovirus 71 (EV-71) is the main etiological agent of hand, foot and mouth disease (HFMD). Recent EV-71 outbreaks in Asia-Pacific were not limited to mild HFMD, but were associated with severe neurological complications such as aseptic meningitis and brainstem encephalitis, which may lead to cardiopulmonary failure and death. The absence of licensed therapeutics for clinical use has intensified research into anti-EV-71 development. This review highlights the potential antiviral agents targeting EV-71 attachment, entry, uncoating, translation, polyprotein processing, virus-induced formation of membranous RNA replication complexes, and RNA-dependent RNA polymerase. The strategies for antiviral development include target-based synthetic compounds, anti-rhinovirus and poliovirus libraries screening, and natural compound libraries screening. Growing knowledge of the EV-71 life cycle will lead to successful development of antivirals. The continued effort to develop antiviral agents for treatment is crucial in the absence of a vaccine. The coupling of antivirals with an effective vaccine will accelerate eradication of the disease.
Similar content being viewed by others

Introduction
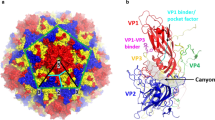
Human enterovirus A71 (EV-71) belongs to the Enterovirus genus within the family of Picornaviridae. The EV-71 genome is a single-stranded, positive sense RNA with approximately 7411 nucleotides, and consists of an open reading frame flanked by 5′ and 3′ untranslated regions (UTRs) [1]. Internal ribosome entry site (IRES)-dependent translation initiates synthesis of the viral polyprotein, which is subsequently cleaved into structural proteins (VP1-VP4) and non-structural proteins (2A-2C and 3A-3D). The RNA genome is enclosed in an icosahedral capsid assembled from 60 copies of each of the four structural proteins [2].
EV-71 was first described in 1969, after its isolation from a two-month-old infant with aseptic meningitis in California, USA. Several EV-71 epidemics with high mortality rates occurred in Bulgaria and Hungary in 1975 and 1978 [3–5], respectively. Since then, many EV-71 outbreaks have been reported in Taiwan [6], Australia [7], Singapore [8], Malaysia [9], China [10–14], Vietnam [15] and Cambodia [16].
EV-71 infections usually manifest as mild hand, foot and mouth disease (HFMD), characterized by fever, mouth ulcers, and vesicles on the palms and feet. Unlike other HFMD-related enteroviruses, EV-71 also causes severe neurological manifestations, such as poliomyelitis-like acute flaccid paralysis and brainstem encephalitis in infants and children below 6 years old [17, 18]. The fatal brainstem encephalitis is characterized by rapid progression of cardiopulmonary failure. Patients with neurological involvement who survive often have permanent neurological sequelae, with delayed neurodevelopment and reduced cognitive function [19, 20].
Similar to the global poliovirus (PV) eradication initiative, an EV-71 vaccine is likely to be the most effective way to control, and hopefully eradicate disease [21, 22]. Several promising EV-71 vaccine candidates are currently under clinical trial [23]. Nevertheless, effective antivirals are still needed for treatment of infected patients with severe disease [21, 22]. This review will highlight the potential targets for EV-71 antivirals as well as recent developments and future prospects of antivirals against EV-71 infections.
Review
EV-71 virus life cycle
Similar to other viruses, EV-71 infection begins with initial attachment to attachment factors present on the cell surface, followed by interaction with entry receptors. EV-71 enters the cells through clathrin-mediated endocytosis and uncoats in the early endosomes. The viral RNA undergoes IRES-dependent translation, and the polyprotein is cleaved by 2A and 3C proteases into structural and non-structural proteins. Non-structural proteins are mainly involved in negative-sense and positive-sense RNA synthesis. The positive-sense viral RNA is then packed into the procapsid, which finally matures into infectious viral particles. Details of the EV-71 replication steps will be discussed according to their therapeutic targets [18, 21, 22].
Therapeutics targeting viral attachment and entry
Virus-host receptor interaction is the first essential event during virus infection. The ability to recognize and bind to specific receptors determines the host range and tissue tropism [24]. Cell surface carbohydrates such as heparan sulfate glycosaminoglycan and sialic acid are often targeted by pathogens as attachment factors. EV-71 uses cell surface heparan sulfate [25] and sialylated glycan [26, 27] as attachment receptors, which could concentrate the virus on the host cell surface and therefore enhance infectivity. Further interaction with entry receptors is required to initiate infection. Two functionally important entry receptors have been identified, scavenger receptor class B2 (SCARB2) and P-selectin glycoprotein ligand-1 (PSGL-1) [28, 29]. SCARB2 is expressed in all cell types and regarded as the major EV-71 entry receptor. At low endosomal pH, SCARB2 is needed to induce viral uncoating [30, 31]. Human SCARB2 transgenic mice infected with EV-71 showed lethal neurological manifestations with pathological features similar to humans and monkeys, suggesting that SCARB2 contributes to its pathogenesis [32, 33]. PSGL-1 is only present on neutrophils and leukocytes. EV-71 binds to PSGL-1 and enters the cells through the caveolar endocytosis pathway [34]. Transgenic mice expressing human PSGL-1 failed to enhance EV-71 infectivity, suggested that PSGL-1 alone does not contribute to its pathogenesis [35].
Since host-receptor interactions are the first event during infection, inhibitors that block this event could act as potential therapeutics. The soluble form of cellular receptors could act as molecular decoys of cell-associated receptors. Soluble SCARB2, PSGL-1, sialic acid and heparin or heparin mimetics have been demonstrated to exhibit inhibitory effects against EV-71 infection in vitro[25, 26, 28, 30, 36]. Highly sulfated suramin and its analog, NF449, exhibited antiviral activity against EV-71 infection [25, 37]. NF449-resistant mutants consist of two mutations in VP1, E98Q and K244R, implying that NF449 inhibited EV-71 infection by binding to the VP1 protein [37]. Similarly, kappa carrageenan, a sulfated polysaccharide from seaweed, also exhibited significant antiviral activity through targeting EV-71 attachment and entry [38]. The mechanism of these soluble decoys is possibly by disruption of the integrity of the EV-71 capsid structure or steric hindrance of receptor interactions.
Receptor antagonists could also be developed as potential antiviral agents. A peptide derived from EV-71 VP1, designated SP40 peptide (Ac-QMRRKVELFTYMRFD-NH2), was found to exhibit significant antiviral activity against different strains of EV-71 by blocking viral attachment to the cell surface heparan sulfate [39]. An anti-heparan sulfate peptide (Ac-MPRRRRIRRRQK-NH2), previously identified by Tiwari et al.[40], also inhibited EV-71 infection [25]. Another antimicrobial peptide, lactoferrin, also exhibited anti-EV-71 properties in vitro and in vivo through blocking viral attachment to the cell surface [41–43].
Therapeutics targeting viral uncoating
The proposed EV-71 uncoating event involves attachment to the entry receptor, triggering a series of conformational changes resulting in A-particle formation that is primed for genome release. A second uncoating event occurs after endocytosis, and an unknown trigger causes RNA expulsion from the A-particles via the 2-fold axis, leaving behind an empty capsid [44]. Formation of the 135S A-particle happens in the presence of SCARB2 receptors and a low pH environment, suggesting that the A-particle is formed in the early endosomes [30, 31]. Uncoating inhibitors (pocket binders) have been intensively studied as antiviral agents against many picornaviruses, including rhinovirus [45], PV [45], echovirus [46] and coxsackievirus [47]. The complex of WIN51711 with the EV-71 hydrophobic pocket underneath the canyon depression has recently been resolved by X-ray crystallography [48]. The key success factor of these uncoating inhibitors is their ability to fit into the VP1 hydrophobic pocket, stabilize the capsid structure, and therefore block the receptor-induced uncoating mechanism [48].
A series of modified WIN compounds including BPROZ-194, BPROZ-112, BPROZ-284, BPROZ-103, BPROZ-299, BPROZ-101, BPROZ-033, and BPROZ-074 were effective against EV-71 infection with IC50 values ranging from 0.8 nM to 1550 nM [49–54]. However, a single point mutation in VP1 V192M was sufficient to confer resistance to BPROZ-194 [51]. Other than modified WIN compounds, the broad spectrum enterovirus inhibitor pleconaril also inhibited EV-71 infection in vitro and in vivo[55, 56]. However, pleconaril failed to inhibit the cytopathic effect induced by a Taiwan 1998 EV-71 isolate [49]. Another group of capsid binders, pyridazinyl oxime ethers chemically derived from pirodavir such as BTA39 and BTA188, significantly inhibited EV-71 infection [57]. Crystallographic studies showed the pirodavir predecessor R61837 complexed with rhinovirus 14 by binding to the hydrophobic pocket underneath the canyon floor, similar to the mechanism of WIN compounds [58]. 4′,6-Dichloroflavan (BW683C), previously identified as an anti-rhinovirus compound, was also effective against EV-71 infection [59, 60]. Mechanistic studies demonstrated that BW683C binds to and stabilizes rhinovirus to heat or acid inactivation, implying that BW682C acts as viral uncoating inhibitor [61–63].
Therapeutics targeting viral RNA translation
EV-71 protein synthesis commences with translation initiation of the cap-independent IRES element at the 5′UTR of the EV-71 genome [64]. IRES is a cis- acting element that forms tertiary RNA structures and requires assistance from IRES-specific trans-acting factors (ITAFs) to recruit other cellular translation machinery to the viral RNA. The EV-71 open reading frame (ORF) is translated into a single polyprotein, which is subsequently processed by virus-encoded proteases 2A and 3C into the structural capsid proteins (VP1-VP4) and the nonstructural proteins (2A-2C and 3A-3D) mainly involved in the replication of the viral RNA [65].
The antisense-mediated mechanism consists of oligonucleotides (8-50 nucleotides in length) that bind to RNA through Watson-Crick base pairing and modulate the function of the targeted RNA [66]. RNA interference (RNAi) involves the cleavage of targeted mRNA through the RNA-induced silencing complex. Small interfering RNA (siRNA) targeting highly conserved regions of 5′UTR [67], VP1, VP2 [68], 2C, 3C, 3D [69, 70], and 3′UTR [69] significantly inhibited EV-71 infection in a dose-dependent manner. In addition, short hairpin RNA (shRNA) was effective against EV-71 infection in vitro and in vivo[70–72]. The use of siRNA in clinical settings is hampered by its short half-life in plasma. Improved siRNA with 2′O methylation and 2′ fluoro modifications have recently been demonstrated against EV-71 infection [67]. However, siRNA also has poor endosomal uptake which limits the clinical application of these siRNAs. Other translation suppressing nucleotides, for example, peptide conjugated phosphodiamidate morpholino oligomers (PPMO) showed promising results in inhibiting PV and coxsackievirus B3 [73, 74]. Unlike siRNA or shRNA, PPMO interacts with targeted RNA, especially the IRES region, and blocks ribosome recruitment and therefore inhibits viral RNA translation [66]. PPMO readily penetrates the cells and is resistant to nuclease degradation. Our unpublished data confirms that PMO are highly effective against EV-71.
Compounds that down-regulate the activity of IRES-dependent translation could potentially be developed into antiviral agents. Quinacrine, which impairs IRES-dependent translation by preventing the interaction between polypyrimidine-tract binding protein (PTB) and IRES, has been demonstrated to act against EV-71 infection [75]. Kaempferol, a flavonoid, was found to inhibit EV-71 IRES activity by altering the composition of ITAFs [76]. Geniposide derived from Fructus gardeniae inhibited EV-71 replication via inhibition of viral IRES activity [77]. Amantadine, a tricyclic symmetric amine previously used against influenza A virus infection, was found to suppress EV-71 IRES translation [78–80].
Therapeutics targeting viral polyprotein processing
Maturation cleavage of polyprotein into different viral proteins is a critical step during EV-71 infection. EV-71 2A and 3C protease are the key proteases that cleave the viral precursor polyprotein into each of the component proteins required for viral replication and packaging. Interestingly, EV-71 2A and 3C proteases suppress type I interferon by targeting mitochondrial anti-viral signaling (MAVS) protein and melanoma differentiation associated gene (MDA-5) viral recognition receptor signaling [81, 82]. Since EV-71 2A and 3C proteases are involved in multiple roles in EV-71 infection and evasion of host innate immunity, they are important potential targets for development of antiviral therapeutics.
A pseudosubstrate, LVLQTM peptide, could inhibit EV-71 infection through binding to the active site of 2A protease [83]. Rupintrivir (AG7088) is an irreversible peptidomimetic inhibitor of human rhinovirus 3C protease, which reached phase 2 clinical trials with promising outcomes [84–89]. Rupintrivir showed significant inhibition of EV-71 infection in vitro and in vivo but with reduced efficacy as compared with human rhinoviruses [90–93]. X-ray crystallography of the complex of EV-71 3C protease with rupintrivir revealed that the half-closed S2 sub-site and the size reduced S1′ pocket of EV-71 3C protease limits the access of the rupintrivir’s P1′ group which contains a lactam ring [94, 95]. A series of 3C protease rupintrivir analogues were designed based on AG7088, with an aldehyde replacement of the α,β-unsaturated ester. Compound 10b significantly inhibited EV-71 infection [96]. An orally bioavailable 3C protease inhibitor, designated as compound 1, also exhibited antiviral activities against multiple rhinovirus serotypes and enteroviruses in vitro[89]. Flavonoids such as fisetin and rutin, have also been identified as 3C protease inhibitors [97].
Therapeutics targeting the membranous viral RNA replication complex and other host factors
The genomic replication of enteroviruses has been shown to occur in membranous compartments in the cytoplasm. The membranous vesicles induced during PV infection have been reported to be associated with autophagy signalling [98, 99]. These compartments resemble the autophagosomes and consist of viral proteins as well as microtubule-associated protein 1 light chain 3-II (LC3-II). LC3-II is the membrane-bound form of LC3 that serves as the marker of autophagy induction [100]. During PV infection, these double-membrane vesicles consist of viral particles that undergo autophagic maturation typically characterized by LC3-II co-localization with lysosomal-associated membrane protein 1 (LAMP1) [100]. Similarly, EV-71 induces autophagy formation in RD and SK-N-SH cells, and association between autophagosome-like vesicles and EV-71 VP1 in neurons of the cervical spinal cords of mice was observed [101]. The authors concluded that autophagic signalling induced by EV-71 is crucial for EV-71 replication. This provides an alternative antiviral strategy for EV-71 to target host factors related to autophagy that are crucial for viral replication.
The discovery of antiviral drugs is mainly based on virus targets. The high replication and mutation rates of enteroviruses may generate resistance to these direct-acting antivirals. Targeting host factors may establish a higher genetic barrier to resistance and can be used in combination with viral inhibitors. The compound GW5074, a Raf-1 inhibitor, has been shown to influence EV-71 viral yield [37, 102]. Activation of the Raf-1/ERK pathway in host cells induces autophagy signalling [103]. The downstream transducer of this pathway, BNIP3 competes with Beclin 1 for binding with Bcl-2 during autophagy induction [104]. GW5074 may impair autophagy activation through the inhibition of the Raf-1/ERK pathway. Thus, the replication of EV-71 that requires autophagosome formation may be inhibited in the presence of the GW5074 compound. Heat shock protein 90 beta (HSP90β), an isomer of HSP90, has been reported to have crucial roles in EV-71 entry and assembly. Geldanamycin (GA) and its analog, 17-allyamino-17-demethoxygeldanamycin (17-AAG), inhibit HSP90β activities and protect hSCARB2 transgenic mice from the challenge with EV-71 [105].
Inhibitors that target host factors such as those involved in cellular autophagy and HSP90β could be used against multiple EV-71 genotypes and enterovirus serotypes, due to their similar pathways of replication [106, 107]. The major drawbacks of these inhibitors that target host factors are specificity and cellular toxicity. Therefore, there is an unmet need to develop specific and non-toxic antivirals that impair the cellular autophagy pathway and HSP90β during EV-71 infection.
The amino acid sequences of the non-structural proteins of EV-71 are highly conserved and have more than 60% similarity to PV. Two hydrophobic regions are found in the 2B viral protein of PV and are pivotal for its viroporin functionality [108]. 2B viroporin mediates the integration of viral protein into the ER membrane and this increases the membrane permeability to promote virus release [108]. A study has reported that EV-71 2B protein might mediate a chloride-dependent current in oocytes. A chloride-dependent current inhibitor, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDS) has been reported to inhibit EV-71 infection in RD cells [109]. The 2C viral protein of PV consists of Walker A, B and C motifs that are homologous to the motifs found in NTP-binding proteins or in members of the helicase superfamily III [110]. An amphipathic helix domain is located at the N-terminal of 2C viral protein that has the function of promoting oligomerization [110]. Recently, two antiviral compounds, metrifudil (N-(2-methylphenyl) methyl adenosine) and N6-benzyladenosine, blocked EV-71 replication via interaction with 2C viral protein or 2BC precursor protein [37]. Mutants resistant to metrifudil had a mutation in the 2C viral protein (E325G), while N6-benzyladenosine-resistant mutants had double mutations at the 2C viral protein (H118Y and I324M) [37]. However, the mechanism of inhibition is yet to be determined. Both MRL-1237 and TBZE-029, derivatives of benzimidazole, exhibit antiviral activity against various enteroviruses, and have been identified to target the picornaviral 2C viral protein [111, 112]. Both of these derivatives may exert potent antiviral activity against EV-71 since EV-71 and PV shared high similarity in all the non-structural proteins. Guanidine hydrochloride is an extensively-studied picornavirus inhibitor [113, 114], which inhibits the replication of PV [115, 116], coxsackieviruses [117], echoviruses, and foot-and-mouth disease virus [118]. Interestingly, guanidine hydrochloride also inhibits EV-71 infection and a single mutation, M193L at the 2C protein was sufficient to confer resistance [119]. This agent is likely to prevent the association of 2C/2BC with host membrane structures during viral replication [120].
The 3A viral protein of PV contains hydrophobic domains that facilitate its binding with membranous vesicles induced during viral RNA replication [121, 122]. A benzimidazole derivative, enviroxime exhibits potent activity against PV and rhinovirus by interacting with 3A viral protein [119]. Strong antiviral effects of enviroxime have been shown against EV-71 [123]. Bifunctional inhibitors AN-12-H5 and AN-23-F6, are enviroxime-like compound that also targets 3A, VP1 and VP3, inhibits EV-71 infection efficiently [124]. However, the precise mechanism of action by enviroxime and AN-12-H5 against EV-71 infection remains unknown. Another compound, TTP-8307, was identified as a potent 3A inhibitor that significantly inhibited CV-A16 infection, with reduced activity against EV-71 [112].
Therapeutics targeting RNA-dependent RNA polymerase (RdRP) complex
The viral RNA replication of enteroviruses begins with the linkage of genomic RNA with the 3B protein (VPg) at the 5′ end to form the uridylylated state of VPg (VPg-pUpU). Additionally, VPg uridylylation is stimulated by the viral precursor protein 3CD [125]. The positive strand of viral RNA is used as a template to synthesize the negative strand, which in turn serves as the template for the synthesis of new positive strands. The synthesis of both positive and negative strands of viral RNA is primed by VPg-pUpU [126]. Nucleotide site 311 of the RNA-dependent RNA polymerase (RdRP) of EV-71 is pivotal for VPg uridylylation and viral RNA synthesis, as mutations here impair the binding of VPg to RdRP, but did not influence normal RdRP activity [127].
Ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxyamine) is a conventional nucleoside analogue that targets the RdRP of picornaviruses [128]. Ribavirin inhibits EV-71 infection with an IC50 of 266 μM, and prevents EV-71 induced paralysis and death in mice [129]. Recently, a piperazine-containing pyrazolo [3,4-d] pyrimidine derivative, DTriP-22, was shown to effectively target the RdRP of EV-71 with IC50 values of 0.15 – 0.98 μM, and suppress the accumulation of both positive and negative strands of viral RNA during EV-71 infection. DTriP-22-resistant mutants had mutations in the RdRP, implying that DTriP-22 interacts with RdRP and inhibits poly (U) elongation activity, but not VPg uridylylation [130].
Conclusion
Figure 1 and Table 1 summarizes all the potential targets of antivirals and lists the recent antiviral agents with significant antiviral activities against EV-71 infection as discussed above. Amongst these drugs, modified WIN compounds are antivirals with the lowest IC50. Only bovine lactoferrin, pleconaril, shRNA, siRNA, rupintrivir, ribavirin and 17-AAG have been tested in vivo. Ribavirin and amantadine are already in clinical use for other viruses, and rupintrivir and pleconaril are in clinical development.

Schematic illustration of EV-71 intracellular infection and summary of the antiviral agents. The antiviral agents are classified according to their mechanism of actions, which include molecular decoys, receptor antagonists, uncoating inhibitors, translation inhibitors, polyprotein processing inhibitors and replication inhibitors.
The availability of a suitable animal model carrying all the required receptors and attachment factors for testing of the antivirals will accelerate the development of antivirals. The clinical use of other antiviral agents has been hampered by the potential adverse effects to the host and emergence of drug resistance mutants. Combination therapy targeting different replication steps of EV-71 infection cycle has shown synergistic activity [131] and could minimize the emergence of antiviral resistance. A new antiviral strategy to screen all licensed drugs against EV-71 infection would be more promising for clinical use. Other newer antivirals that act as immunomodulators and lethal mutagens offer a new strategy for development of antivirals. With the endemic and epidemic nature of EV-71, the continued efforts to develop antiviral agents for prophylaxis or treatment are crucial in the absence of a vaccine. Together with an effective vaccine, eradication of EV-71 is anticipated.
Abbreviations
- EV-71:
-
Enterovirus 71
- HFMD:
-
Hand, foot and mouth disease
- IRES:
-
Internal ribosome entry site
- ITAF:
-
IRES-specific trans-acting factor
- MAVS:
-
Mitochondrial anti-viral signaling
- MDA-5:
-
Melanoma differentiation associated gene
- ORF:
-
Open reading frame
- PV:
-
Poliovirus
- RdRP:
-
RNA-dependent RNA polymerase
- VPg:
-
Viral protein genome-linked.
References
Brown BA, Pallansch MA: Complete nucleotide sequence of enterovirus 71 is distinct from poliovirus. Virus Res. 1995, 39: 195-205. 10.1016/0168-1702(95)00087-9.
Plevka P, Perera R, Cardosa J, Kuhn RJ, Rossmann MG: Crystal structure of human enterovirus 71. Science. 2012, 336: 1274-10.1126/science.1218713.
Chumakov M, Voroshilova M, Shindarov L, Lavrova I, Gracheva L, Koroleva G, Vasilenko S, Brodvarova I, Nikolova M, Gyurova S: Enterovirus 71 isolated from cases of epidemic poliomyelitis-like disease in Bulgaria. Arch Virol. 1979, 60: 329-340. 10.1007/BF01317504.
Shindarov LM, Chumakov MP, Voroshilova MK, Bojinov S, Vasilenko SM, Iordanov I, Kirov ID, Kamenov E, Leshchinskaya EV, Mitov G: Epidemiological, clinical, and pathomorphological characteristics of epidemic poliomyelitis-like disease caused by enterovirus 71. J Hyg Epidemiol Microbiol Immunol. 1979, 23: 284-295.
Nagy G, Takatsy S, Kukan E, Mihaly I, Domok I: Virological diagnosis of enterovirus type 71 infections: experiences gained during an epidemic of acute CNS diseases in Hungary in 1978. Arch Virol. 1982, 71: 217-227. 10.1007/BF01314873.
Huang KY, Zhang X, Chung PH, Tsao KC, Lin TY, Su LH, Chiu CH: Enterovirus 71 in Taiwan, 2004-2006: epidemiological and virological features. Scand J Infect Dis. 2008, 40: 571-574. 10.1080/00365540701799359.
Brown BA, Oberste MS, Alexander JP, Kennett ML, Pallansch MA: Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virol. 1999, 73: 9969-9975.
Chan KP, Goh KT, Chong CY, Teo ES, Lau G, Ling AE: Epidemic hand, foot and mouth disease caused by human enterovirus 71, Singapore. Emerg Infect Dis. 2003, 9: 78-85. 10.3201/eid1301.020112.
AbuBakar S, Chee HY, Al-Kobaisi MF, Xiaoshan J, Chua KB, Lam SK: Identification of enterovirus 71 isolates from an outbreak of hand, foot and mouth disease (HFMD) with fatal cases of encephalomyelitis in Malaysia. Virus Res. 1999, 61: 1-9. 10.1016/S0168-1702(99)00019-2.
Tan X, Huang X, Zhu S, Chen H, Yu Q, Wang H, Huo X, Zhou J, Wu Y, Yan D: The persistent circulation of enterovirus 71 in People’s Republic of China: causing emerging nationwide epidemics since 2008. PLoS One. 2011, 6: e25662-10.1371/journal.pone.0025662.
Zhang Y, Tan XJ, Wang HY, Yan DM, Zhu SL, Wang DY, Ji F, Wang XJ, Gao YJ, Chen L: An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J Clin Virol. 2009, 44: 262-267. 10.1016/j.jcv.2009.02.002.
Zhang Y, Wang J, Guo W, Wang H, Zhu S, Wang D, Bai R, Li X, Yan D, Zhu Z: Emergence and transmission pathways of rapidly evolving evolutionary branch C4a strains of human enterovirus 71 in the Central Plain of China. PLoS One. 2011, 6: e27895-10.1371/journal.pone.0027895.
Zhang Y, Zhu Z, Yang W, Ren J, Tan X, Wang Y, Mao N, Xu S, Zhu S, Cui A: An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of China. Virol J. 2010, 7: 94-10.1186/1743-422X-7-94.
Zhu RN, Qian Y, Deng J, Xing JF, Zhao LQ, Wang F, Liao B, Ren XX, Li Y, Zhang Q, Li J: Study on the association of hand, foot and mouth disease and enterovirus 71/CA16 among children in Beijing, 2007. Zhonghua Liu Xing Bing Xue Za Zhi. 2007, 28: 1004-1008.
le Thoa PK, Chiang PS, Khanh TH, Luo ST, Dan TN, Wang YF, Thuong TC, Chung WY, Hung NT, Wang JR: Genetic and antigenic characterization of enterovirus 71 in Ho Chi Minh city, Vietnam, 2011. PLoS One. 2013, 8: e69895-10.1371/journal.pone.0069895.
Biswas T: Enterovirus 71 causes hand, foot and mouth disease outbreak in Cambodia. Natl Med J India. 2012, 25: 316-
Ooi MH, Wong SC, Lewthwaite P, Cardosa MJ, Solomon T: Clinical features, diagnosis, and management of enterovirus 71. Lancet Neurol. 2010, 9: 1097-1105. 10.1016/S1474-4422(10)70209-X.
Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH: Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis. 2010, 10: 778-790. 10.1016/S1473-3099(10)70194-8.
Chang LY, Lin TY, Hsu KH, Huang YC, Lin KL, Hsueh C, Shih SR, Ning HC, Hwang MS, Wang HS, Lee CY: Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet. 1999, 354: 1682-1686. 10.1016/S0140-6736(99)04434-7.
Chang LY, Huang LM, Gau SS, Wu YY, Hsia SH, Fan TY, Lin KL, Huang YC, Lu CY, Lin TY: Neurodevelopment and cognition in children after enterovirus 71 infection. N Engl J Med. 2007, 356: 1226-1234. 10.1056/NEJMoa065954.
Shang LQ, Xu MY, Yin Z: Antiviral drug discovery for the treatment of enterovirus 71 infections. Antiviral Res. 2013, 97: 183-194. 10.1016/j.antiviral.2012.12.005.
Kuo RL, Shih SR: Strategies to develop antivirals against enterovirus 71. Virol J. 2013, 10: 28-10.1186/1743-422X-10-28.
Chong P, Hsieh SY, Liu CC, Chou AH, Chang JY, Wu SC, Liu SJ, Chow YH, Su IJ, Klein M: Production of EV71 vaccine candidates. Hum Vaccin Immunother. 2012, 8: 1775-1783. 10.4161/hv.21739.
Haywood AM: Virus receptors: binding, adhesion strengthening, and changes in viral structure. J Virol. 1994, 68: 1-5.
Tan CW, Poh CL, Sam IC, Chan YF: Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J Virol. 2013, 87: 611-620. 10.1128/JVI.02226-12.
Yang B, Chuang H, Yang KD: Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol J. 2009, 6: 141-10.1186/1743-422X-6-141.
Su PY, Liu YT, Chang HY, Huang SW, Wang YF, Yu CK, Wang JR, Chang CF: Cell surface sialylation affects binding of enterovirus 71 to rhabdomyosarcoma and neuroblastoma cells. BMC Microbiol. 2012, 12: 162-10.1186/1471-2180-12-162.
Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S: Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat Med. 2009, 15: 798-801. 10.1038/nm.1992.
Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H: Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat Med. 2009, 15: 794-797. 10.1038/nm.1961.
Yamayoshi S, Ohka S, Fujii K, Koike S: Functional comparison of SCARB2 and PSGL1 as receptors for enterovirus 71. J Virol. 2013, 87: 3335-3347. 10.1128/JVI.02070-12.
Chen P, Song Z, Qi Y, Feng X, Xu N, Sun Y, Wu X, Yao X, Mao Q, Li X: Molecular determinants of enterovirus 71 viral entry: cleft around GLN-172 on VP1 protein interacts with variable region on scavenge receptor B 2. J Biol Chem. 2012, 287: 6406-6420. 10.1074/jbc.M111.301622.
Lin YW, Yu SL, Shao HY, Lin HY, Liu CC, Hsiao KN, Chitra E, Tsou YL, Chang HW, Sia C: Human SCARB2 transgenic mice as an infectious animal model for enterovirus 71. PLoS One. 2013, 8: e57591-10.1371/journal.pone.0057591.
Fujii K, Nagata N, Sato Y, Ong KC, Wong KT, Yamayoshi S, Shimanuki M, Shitara H, Taya C, Koike S: Transgenic mouse model for the study of enterovirus 71 neuropathogenesis. Proc Natl Acad Sci U S A. 2013, 110: 14753-14758. 10.1073/pnas.1217563110.
Lin HY, Yang YT, Yu SL, Hsiao KN, Liu CC, Sia C, Chow YH: Caveolar endocytosis is required for human PSGL-1-mediated enterovirus 71 infection. J Virol. 2013, 87: 9064-9076. 10.1128/JVI.00573-13.
Liu J, Dong W, Quan X, Ma C, Qin C, Zhang L: Transgenic expression of human P-selectin glycoprotein ligand-1 is not sufficient for enterovirus 71 infection in mice. Arch Virol. 2012, 157: 539-543. 10.1007/s00705-011-1198-2.
Pourianfar HR, Poh CL, Fecondo J, Grollo L: In vitro evaluation of the antiviral activity of heparan sulphate mimetic compounds against enterovirus 71. Virus Res. 2012, 169: 22-29. 10.1016/j.virusres.2012.06.025.
Arita M, Wakita T, Shimizu H: Characterization of pharmacologically active compounds that inhibit poliovirus and enterovirus 71 infectivity. J Gen Virol. 2008, 89: 2518-2530. 10.1099/vir.0.2008/002915-0.
Chiu YH, Chan YL, Tsai LW, Li TL, Wu CJ: Prevention of human enterovirus 71 infection by kappa carrageenan. Antiviral Res. 2012, 95: 128-134. 10.1016/j.antiviral.2012.05.009.
Tan CW, Chan YF, Sim KM, Tan EL, Poh CL: Inhibition of enterovirus 71 (EV-71) infections by a novel antiviral peptide derived from EV-71 capsid protein VP1. PLoS One. 2012, 7: e34589-10.1371/journal.pone.0034589.
Tiwari V, Liu J, Valyi-Nagy T, Shukla D: Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J Biol Chem. 2011, 286: 25406-25415. 10.1074/jbc.M110.201103.
Chen HL, Wang LC, Chang CH, Yen CC, Cheng WT, Wu SC, Hung CM, Kuo MF, Chen CM: Recombinant porcine lactoferrin expressed in the milk of transgenic mice protects neonatal mice from a lethal challenge with enterovirus type 71. Vaccine. 2008, 26: 891-898. 10.1016/j.vaccine.2007.12.013.
Lin TY, Chu C, Chiu CH: Lactoferrin inhibits enterovirus 71 infection of human embryonal rhabdomyosarcoma cells in vitro. J Infect Dis. 2002, 186: 1161-1164. 10.1086/343809.
Weng TY, Chen LC, Shyu HW, Chen SH, Wang JR, Yu CK, Lei HY, Yeh TM: Lactoferrin inhibits enterovirus 71 infection by binding to VP1 protein and host cells. Antiviral Res. 2005, 67: 31-37. 10.1016/j.antiviral.2005.03.005.
Shingler KL, Yoder JL, Carnegie MS, Ashley RE, Makhov AM, Conway JF, Hafenstein S: The enterovirus 71 A-particle forms a gateway to allow genome release: a cryoEM study of picornavirus uncoating. PLoS Pathog. 2013, 9: e1003240-10.1371/journal.ppat.1003240.
Fox MP, Otto MJ, McKinlay MA: Prevention of rhinovirus and poliovirus uncoating by WIN 51711, a new antiviral drug. Antimicrob Agents Chemother. 1986, 30: 110-116. 10.1128/AAC.30.1.110.
McKinlay MA, Frank JA, Benziger DP, Steinberg BA: Use of WIN 51711 to prevent echovirus type 9-induced paralysis in suckling mice. J Infect Dis. 1986, 154: 676-681. 10.1093/infdis/154.4.676.
See DM, Tilles JG: Treatment of coxsackievirus A9 myocarditis in mice with WIN 54954. Antimicrob Agents Chemother. 1992, 36: 425-428. 10.1128/AAC.36.2.425.
Plevka P, Perera R, Yap ML, Cardosa J, Kuhn RJ, Rossmann MG: Structure of human enterovirus 71 in complex with a capsid-binding inhibitor. Proc Natl Acad Sci U S A. 2013, 110: 5463-5467. 10.1073/pnas.1222379110.
Shia KS, Li WT, Chang CM, Hsu MC, Chern JH, Leong MK, Tseng SN, Lee CC, Lee YC, Chen SJ: Design, synthesis, and structure-activity relationship of pyridyl imidazolidinones: a novel class of potent and selective human enterovirus 71 inhibitors. J Med Chem. 2002, 45: 1644-1655. 10.1021/jm010536a.
Shih SR, Chen SJ, Hakimelahi GH, Liu HJ, Tseng CT, Shia KS: Selective human enterovirus and rhinovirus inhibitors: an overview of capsid-binding and protease-inhibiting molecules. Med Res Rev. 2004, 24: 449-474. 10.1002/med.10067.
Shih SR, Tsai MC, Tseng SN, Won KF, Shia KS, Li WT, Chern JH, Chen GW, Lee CC, Lee YC: Mutation in enterovirus 71 capsid protein VP1 confers resistance to the inhibitory effects of pyridyl imidazolidinone. Antimicrob Agents Chemother. 2004, 48: 3523-3529. 10.1128/AAC.48.9.3523-3529.2004.
Chen TC, Liu SC, Huang PN, Chang HY, Chern JH, Shih SR: Antiviral activity of pyridyl imidazolidinones against enterovirus 71 variants. J Biomed Sci. 2008, 15: 291-300. 10.1007/s11373-007-9228-5.
Chern JH, Shia KS, Hsu TA, Tai CL, Lee CC, Lee YC, Chang CS, Tseng SN, Shih SR: Design, synthesis, and structure-activity relationships of pyrazolo [3,4-d]pyrimidines: a novel class of potent enterovirus inhibitors. Bioorg Med Chem Lett. 2004, 14: 2519-2525. 10.1016/j.bmcl.2004.02.092.
Chang CS, Lin YT, Shih SR, Lee CC, Lee YC, Tai CL, Tseng SN, Chern JH: Design, synthesis, and antipicornavirus activity of 1-[5-(4-arylphenoxy)alkyl]-3-pyridin-4-ylimidazolidin-2-one derivatives. J Med Chem. 2005, 48: 3522-3535. 10.1021/jm050033v.
Pevear DC, Tull TM, Seipel ME, Groarke JM: Activity of pleconaril against enteroviruses. Antimicrob Agents Chemother. 1999, 43: 2109-2115.
Zhang G, Zhou F, Gu B, Ding C, Feng D, Xie F, Wang J, Zhang C, Cao Q, Deng Y: In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch Virol. 2012, 157: 669-679. 10.1007/s00705-011-1222-6.
Barnard DL, Hubbard VD, Smee DF, Sidwell RW, Watson KG, Tucker SP, Reece PA: In vitro activity of expanded-spectrum pyridazinyl oxime ethers related to pirodavir: novel capsid-binding inhibitors with potent antipicornavirus activity. Antimicrob Agents Chemother. 2004, 48: 1766-1772. 10.1128/AAC.48.5.1766-1772.2004.
Chapman MS, Minor I, Rossmann MG, Diana GD, Andries K: Human rhinovirus 14 complexed with antiviral compound R 61837. J Mol Biol. 1991, 217: 455-463. 10.1016/0022-2836(91)90749-V.
Genovese D, Conti C, Tomao P, Desideri N, Stein ML, Catone S, Fiore L: Effect of chloro-, cyano-, and amidino-substituted flavanoids on enterovirus infection in vitro. Antiviral Res. 1995, 27: 123-136. 10.1016/0166-3542(95)00088-4.
Conti C, Mastromarino P, Sgro R, Desideri N: Anti-picornavirus activity of synthetic flavon-3-yl esters. Antivir Chem Chemother. 1998, 9: 511-515.
Bauer DJ, Selway JW, Batchelor JF, Tisdale M, Caldwell IC, Young DA: 4′,6-Dichloroflavan (BW683C), a new anti-rhinovirus compound. Nature. 1981, 292: 369-370. 10.1038/292369a0.
Tisdale M, Selway JW: Inhibition of an early stage of rhinovirus replication by dichloroflavan (BW683C). J Gen Virol. 1983, 64: 795-803. 10.1099/0022-1317-64-4-795.
Tisdale M, Selway JW: Effect of dichloroflavan (BW683C) on the stability and uncoating of rhinovirus type 1B. J Antimicrob Chemother. 1984, 14 (Suppl A): 97-105. 10.1093/jac/14.suppl_A.97.
Thompson SR, Sarnow P: Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology. 2003, 315: 259-266. 10.1016/S0042-6822(03)00544-0.
Lin JY, Chen TC, Weng KF, Chang SC, Chen LL, Shih SR: Viral and host proteins involved in picornavirus life cycle. J Biomed Sci. 2009, 16: 103-10.1186/1423-0127-16-103.
Kole R, Krainer AR, Altman S: RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012, 11: 125-140.
Deng JX, Nie XJ, Lei YF, Ma CF, Xu DL, Li B, Xu ZK, Zhang GC: The highly conserved 5′ untranslated region as an effective target towards the inhibition of enterovirus 71 replication by unmodified and appropriate 2′- modified siRNAs. J Biomed Sci. 2012, 19: 73-10.1186/1423-0127-19-73.
Wu Z, Yang F, Zhao R, Zhao L, Guo D, Jin Q: Identification of small interfering RNAs which inhibit the replication of several enterovirus 71 strains in China. J Virol Methods. 2009, 159: 233-238. 10.1016/j.jviromet.2009.04.002.
Sim AC, Luhur A, Tan TM, Chow VT, Poh CL: RNA interference against enterovirus 71 infection. Virology. 2005, 341: 72-79. 10.1016/j.virol.2005.06.047.
Tan EL, Tan TM, Tak Kwong Chow V, Poh CL: Inhibition of enterovirus 71 in virus-infected mice by RNA interference. Mol Ther. 2007, 15: 1931-1938. 10.1038/sj.mt.6300287.
Tan EL, Tan TM, Chow VT, Poh CL: Enhanced potency and efficacy of 29-mer shRNAs in inhibition of enterovirus 71. Antiviral Res. 2007, 74: 9-15. 10.1016/j.antiviral.2007.01.004.
Lu WW, Hsu YY, Yang JY, Kung SH: Selective inhibition of enterovirus 71 replication by short hairpin RNAs. Biochem Biophys Res Commun. 2004, 325: 494-499. 10.1016/j.bbrc.2004.10.062.
Stone JK, Rijnbrand R, Stein DA, Ma Y, Yang Y, Iversen PL, Andino R: A morpholino oligomer targeting highly conserved internal ribosome entry site sequence is able to inhibit multiple species of picornavirus. Antimicrob Agents Chemother. 2008, 52: 1970-1981. 10.1128/AAC.00011-08.
Yuan J, Stein DA, Lim T, Qiu D, Coughlin S, Liu Z, Wang Y, Blouch R, Moulton HM, Iversen PL, Yang D: Inhibition of coxsackievirus B3 in cell cultures and in mice by peptide-conjugated morpholino oligomers targeting the internal ribosome entry site. J Virol. 2006, 80: 11510-11519. 10.1128/JVI.00900-06.
Wang J, Du J, Wu Z, Jin Q: Quinacrine impairs enterovirus 71 RNA replication by preventing binding of polypyrimidine-tract binding protein with internal ribosome entry sites. PLoS One. 2013, 8: e52954-10.1371/journal.pone.0052954.
Tsai FJ, Lin CW, Lai CC, Lan YC, Lai CH, Hung CH, Hsueh KC, Lin TH, Chang HC, Wan L: Kaempferol inhibits enterovirus 71 replication and internal ribosome entry site (IRES) activity through FUBP and HNRP proteins. Food Chem. 2011, 128: 312-322. 10.1016/j.foodchem.2011.03.022.
Lin YJ, Lai CC, Lai CH, Sue SC, Lin CW, Hung CH, Lin TH, Hsu WY, Huang SM, Hung YL: Inhibition of enterovirus 71 infections and viral IRES activity by Fructus gardeniae and geniposide. Eur J Med Chem. 2013, 62: 206-213.
Chen YJ, Zeng SJ, Hsu JT, Horng JT, Yang HM, Shih SR, Chu YT, Wu TY: Amantadine as a regulator of internal ribosome entry site. Acta Pharmacol Sin. 2008, 29: 1327-1333. 10.1111/j.1745-7254.2008.00876.x.
Davies WL, Grunert RR, Hoffmann CE: Influenza virus growth and antibody response in amantadine-treated mice. J Immunol. 1965, 95: 1090-1094.
Hoffmann CE, Neumayer EM, Haff RF, Goldsby RA: Mode of action of the antiviral activity of amantadine in tissue culture. J Bacteriol. 1965, 90: 623-628.
Wang B, Xi X, Lei X, Zhang X, Cui S, Wang J, Jin Q, Zhao Z: Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9: e1003231-10.1371/journal.ppat.1003231.
Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J: Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J Virol. 2013, 87: 1690-1698. 10.1128/JVI.01855-12.
Falah N, Montserret R, Lelogeais V, Schuffenecker I, Lina B, Cortay JC, Violot S: Blocking human enterovirus 71 replication by targeting viral 2A protease. J Antimicrob Chemother. 2012, 67: 2865-2869. 10.1093/jac/dks304.
Witherell G: AG-7088 Pfizer. Curr Opin Investig Drugs. 2000, 1: 297-302.
Binford SL, Maldonado F, Brothers MA, Weady PT, Zalman LS, Meador JW, Matthews DA, Patick AK: Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob Agents Chemother. 2005, 49: 619-626. 10.1128/AAC.49.2.619-626.2005.
Binford SL, Weady PT, Maldonado F, Brothers MA, Matthews DA, Patick AK: In vitro resistance study of rupintrivir, a novel inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 2007, 51: 4366-4373. 10.1128/AAC.00905-07.
Patick AK: Rhinovirus chemotherapy. Antiviral Res. 2006, 71: 391-396. 10.1016/j.antiviral.2006.03.011.
Patick AK, Binford SL, Brothers MA, Jackson RL, Ford CE, Diem MD, Maldonado F, Dragovich PS, Zhou R, Prins TJ: In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 1999, 43: 2444-2450.
Patick AK, Brothers MA, Maldonado F, Binford S, Maldonado O, Fuhrman S, Petersen A, Smith GJ, Zalman LS, Burns-Naas LA, Tran JQ: In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother. 2005, 49: 2267-2275. 10.1128/AAC.49.6.2267-2275.2005.
Zhang XN, Song ZG, Jiang T, Shi BS, Hu YW, Yuan ZH: Rupintrivir is a promising candidate for treating severe cases of Enterovirus-71 infection. World J Gastroenterol. 2010, 16: 201-209. 10.3748/wjg.v16.i2.201.
Hung HC, Wang HC, Shih SR, Teng IF, Tseng CP, Hsu JT: Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J Infect Dis. 2011, 203: 1784-1790. 10.1093/infdis/jir174.
Lu G, Qi J, Chen Z, Xu X, Gao F, Lin D, Qian W, Liu H, Jiang H, Yan J, Gao GF: Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J Virol. 2011, 85: 10319-10331. 10.1128/JVI.00787-11.
Zhang X, Song Z, Qin B, Chen L, Hu Y, Yuan Z: Rupintrivir is a promising candidate for treating severe cases of enterovirus-71 infection: evaluation of antiviral efficacy in a murine infection model. Antiviral Res. 2013, 97: 264-269. 10.1016/j.antiviral.2012.12.029.
Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang M, Jin Q, Cui S: Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol. 2011, 85: 10021-10030. 10.1128/JVI.05107-11.
Wu C, Cai Q, Chen C, Li N, Peng X, Cai Y, Yin K, Chen X, Wang X, Zhang R: Structures of enterovirus 71 3C proteinase (strain E2004104-TW-CDC) and its complex with rupintrivir. Acta Crystallogr D Biol Crystallogr. 2013, 69: 866-871. 10.1107/S0907444913002862.
Kuo CJ, Shie JJ, Fang JM, Yen GR, Hsu JT, Liu HG, Tseng SN, Chang SC, Lee CY, Shih SR, Liang PH: Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem. 2008, 16: 7388-7398. 10.1016/j.bmc.2008.06.015.
Lin YJ, Chang YC, Hsiao NW, Hsieh JL, Wang CY, Kung SH, Tsai FJ, Lan YC, Lin CW: Fisetin and rutin as 3C protease inhibitors of enterovirus A71. J Virol Methods. 2012, 182: 93-98. 10.1016/j.jviromet.2012.03.020.
Rust RC, Landmann L, Gosert R, Tang BL, Hong W, Hauri HP, Egger D, Bienz K: Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J Virol. 2001, 75: 9808-9818. 10.1128/JVI.75.20.9808-9818.2001.
Egger D, Teterina N, Ehrenfeld E, Bienz K: Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J Virol. 2000, 74: 6570-6580. 10.1128/JVI.74.14.6570-6580.2000.
Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K: Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3: e156-10.1371/journal.pbio.0030156.
Huang SC, Chang CL, Wang PS, Tsai Y, Liu HS: Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J Med Virol. 2009, 81: 1241-1252. 10.1002/jmv.21502.
Arita M, Wakita T, Shimizu H: Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J Gen Virol. 2009, 90: 1869-1879. 10.1099/vir.0.012096-0.
Shima Y, Okamoto T, Aoyama T, Yasura K, Ishibe T, Nishijo K, Shibata KR, Kohno Y, Fukiage K, Otsuka S: In vitro transformation of mesenchymal stem cells by oncogenic H-rasVal12. Biochem Biophys Res Commun. 2007, 353: 60-66. 10.1016/j.bbrc.2006.11.137.
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM: Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009, 29: 2570-2581. 10.1128/MCB.00166-09.
Tsou YL, Lin YW, Chang HW, Lin HY, Shao HY, Yu SL, Liu CC, Chitra E, Sia C, Chow YH: Heat shock protein 90: role in enterovirus 71 entry and assembly and potential target for therapy. PLoS One. 2013, 8: e77133-10.1371/journal.pone.0077133.
Pfefferle S, Schopf J, Kogl M, Friedel CC, Muller MA, Carbajo-Lozoya J, Stellberger T, von Dall’Armi E, Herzog P, Kallies S: The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011, 7: e1002331-10.1371/journal.ppat.1002331.
Marcellin P, Horsmans Y, Nevens F, Grange JD, Bronowicki JP, Vetter D, Purdy S, Garg V, Bengtsson L, McNair L, Alam J: Phase 2 study of the combination of merimepodib with peginterferon-alpha2b, and ribavirin in nonresponders to previous therapy for chronic hepatitis C. J Hepatol. 2007, 47: 476-483. 10.1016/j.jhep.2007.03.028.
Martinez-Gil L, Bano-Polo M, Redondo N, Sanchez-Martinez S, Nieva JL, Carrasco L, Mingarro I: Membrane integration of poliovirus 2B viroporin. J Virol. 2011, 85: 11315-11324. 10.1128/JVI.05421-11.
Xie S, Wang K, Yu W, Lu W, Xu K, Wang J, Ye B, Schwarz W, Jin Q, Sun B: DIDS blocks a chloride-dependent current that is mediated by the 2B protein of enterovirus 71. Cell Res. 2011, 21: 1271-1275. 10.1038/cr.2011.112.
Adams P, Kandiah E, Effantin G, Steven AC, Ehrenfeld E: Poliovirus 2C protein forms homo-oligomeric structures required for ATPase activity. J Biol Chem. 2009, 284: 22012-22021. 10.1074/jbc.M109.031807.
Shimizu H, Agoh M, Agoh Y, Yoshida H, Yoshii K, Yoneyama T, Hagiwara A, Miyamura T: Mutations in the 2C region of poliovirus responsible for altered sensitivity to benzimidazole derivatives. J Virol. 2000, 74: 4146-4154. 10.1128/JVI.74.9.4146-4154.2000.
De Palma AM, Thibaut HJ, van der Linden L, Lanke K, Heggermont W, Ireland S, Andrews R, Arimilli M, Al-Tel TH, De Clercq E: Mutations in the nonstructural protein 3A confer resistance to the novel enterovirus replication inhibitor TTP-8307. Antimicrob Agents Chemother. 2009, 53: 1850-1857. 10.1128/AAC.00934-08.
Caliguiri LA, Tamm I: Distribution and translation of poliovirus RNA in guanidine-treated cells. Virology. 1968, 36: 223-231. 10.1016/0042-6822(68)90139-6.
Caliguiri LA, Tamm I: Action of guanidine on the replication of poliovirus RNA. Virology. 1968, 35: 408-417. 10.1016/0042-6822(68)90219-5.
Loddo B, Ferrari W, Brotzu G, Spanedda A: In vitro inhibition of infectivity of polio viruses by guanidine. Nature. 1962, 193: 97-98. 10.1038/193097a0.
Rightsel WA, Dice JR, Mc AR, Timm EA, Mc LI, Dixon GJ, Schabel FM: Antiviral effect of guanidine. Science. 1961, 134: 558-559. 10.1126/science.134.3478.558.
Herrmann EC, Herrmann JA, Delong DC: Prevention of death in mice infected with coxsackievirus A16 using guanidine HCl mixed with substituted benzimidazoles. Antiviral Res. 1982, 2: 339-346. 10.1016/0166-3542(82)90003-1.
Saunders K, King AM, McCahon D, Newman JW, Slade WR, Forss S: Recombination and oligonucleotide analysis of guanidine-resistant foot-and-mouth disease virus mutants. J Virol. 1985, 56: 921-929.
Sadeghipour S, Bek EJ, McMinn PC: Selection and characterisation of guanidine-resistant mutants of human enterovirus 71. Virus Res. 2012, 169: 72-79. 10.1016/j.virusres.2012.07.005.
Bienz K, Egger D, Troxler M, Pasamontes L: Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J Virol. 1990, 64: 1156-1163.
Taylor MP, Burgon TB, Kirkegaard K, Jackson WT: Role of microtubules in extracellular release of poliovirus. J Virol. 2009, 83: 6599-6609. 10.1128/JVI.01819-08.
Wessels E, Duijsings D, Notebaart RA, Melchers WJ, van Kuppeveld FJ: A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J Virol. 2005, 79: 5163-5173. 10.1128/JVI.79.8.5163-5173.2005.
De Palma AM, Vliegen I, De Clercq E, Neyts J: Selective inhibitors of picornavirus replication. Med Res Rev. 2008, 28: 823-884. 10.1002/med.20125.
Arita M, Takebe Y, Wakita T, Shimizu H: A bifunctional anti-enterovirus compound that inhibits replication and the early stage of enterovirus 71 infection. J Gen Virol. 2010, 91: 2734-2744. 10.1099/vir.0.023374-0.
Pathak HB, Arnold JJ, Wiegand PN, Hargittai MR, Cameron CE: Picornavirus genome replication: assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J Biol Chem. 2007, 282: 16202-16213. 10.1074/jbc.M610608200.
Paul AV, Rieder E, Kim DW, van Boom JH, Wimmer E: Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J Virol. 2000, 74: 10359-10370. 10.1128/JVI.74.22.10359-10370.2000.
Sun Y, Wang Y, Shan C, Chen C, Xu P, Song M, Zhou H, Yang C, Xu W, Shi PY: Enterovirus-71 VPg uridylylation uses a two-molecular mechanism of 3Dpol. J Virol. 2012, 86: 13662-13671. 10.1128/JVI.01712-12.
Graci JD, Too K, Smidansky ED, Edathil JP, Barr EW, Harki DA, Galarraga JE, Bollinger JM, Peterson BR, Loakes D: Lethal mutagenesis of picornaviruses with N-6-modified purine nucleoside analogues. Antimicrob Agents Chemother. 2008, 52: 971-979. 10.1128/AAC.01056-07.
Li ZH, Li CM, Ling P, Shen FH, Chen SH, Liu CC, Yu CK: Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J Infect Dis. 2008, 197: 854-857. 10.1086/527326.
Chen TC, Chang HY, Lin PF, Chern JH, Hsu JT, Chang CY, Shih SR: Novel antiviral agent DTriP-22 targets RNA-dependent RNA polymerase of enterovirus 71. Antimicrob Agents Chemother. 2009, 53: 2740-2747. 10.1128/AAC.00101-09.
Thibaut HJ, Leyssen P, Puerstinger G, Muigg A, Neyts J, De Palma AM: Towards the design of combination therapy for the treatment of enterovirus infections. Antiviral Res. 2011, 90: 213-217. 10.1016/j.antiviral.2011.03.187.
Acknowledgement
This work was supported by the High Impact Research grant (UM.C/625/1/HIR/MOHE/MED/41), postgraduate research grants (PV013-2012A and PG114-2012B), and University Malaya Research Grant Scheme (RG522-13HTM) from University Malaya; and the Fundamental Research Grant Scheme (FP015-2012A) and Exploratory Research Grant Scheme (ER017-2013A) from the Ministry of Education, Malaysia.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CWT and YFC have a pending patent on SP40 peptide.
Authors’ contributions
CWT, JKFL, ICS and YFC drafted the manuscript. All authors read and approved the final manuscript.
Chee Wah Tan contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tan, C.W., Lai, J.K.F., Sam, IC. et al. Recent developments in antiviral agents against enterovirus 71 infection. J Biomed Sci 21, 14 (2014). https://doi.org/10.1186/1423-0127-21-14
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-21-14