
Abstract
Background
Beijing sub-pedigree 2 (BSP2) and T sub-lineage 6 (TSL6) are two clades belonging to Beijing and T family of Mycobacterium tuberculosis (MTB), respectively, defined by Bayesian population structure analysis based on 24-loci mycobacterial interspersed repetitive unit-variable number of tandem repeats (MIRU-VNTR). Globally, over 99% of BSP2 and 89% of TSL6 isolates were distributed in Chongqing, suggesting their possible local adaptive evolution. The objective of this paper is to explore whether BSP2 and TSL6 originated by their local adaptive evolution from the specific isolates of Beijing and T families in Chongqing.
Methods
The genotyping data of 16 090 MTB isolates were collected from laboratory collection, published literatures and SITVIT database before subjected to Bayesian population structure analysis based on 24-loci MIRU-VNTR. Spacer Oligonucleotide Forest (Spoligoforest) and 24-loci MIRU-VNTR-based minimum spanning tree (MST) were used to explore their phylogenetic pathways, with Bayesian demographic analysis for exploring the recent demographic change of TSL6.
Results
Phylogenetic analysis suggested that BSP2 and TSL6 in Chongqing may evolve from BSP4 and TSL5, respectively, which were locally predominant in Tibet and Jiangsu, respectively. Spoligoforest showed that Beijing and T families were genetically distant, while the convergence of the MIRU-VNTR pattern of BSP2 and TSL6 was revealed by WebLogo. The demographic analysis concluded that the recent demographic change of TSL6 might take 111.25 years.
Conclusions
BSP2 and TSL6 clades might originate from BSP4 and TSL5, respectively, by their local adaptive evolution in Chongqing. Our study suggests MIRU-VNTR be combined with other robust markers for a more comprehensive genotyping approach, especially for families of clades with the same MIRU-VNTR pattern.
Similar content being viewed by others


Background
Mycobacterium tuberculosis (MTB) and the other members of the M. tuberculosis complex (MTBC), leading to tuberculosis (TB) in animals and human, have caused estimated over 10 million new infections and 1.24 million deaths in 2018 [1]. There is a strong association between poverty and ill-health [2], and globally, developing countries and low-income countries are the major regions where TB occurs, with the top eight TB high-burden countries accounting for two-thirds of cases worldwide. Among them, China has the second highest burden of TB (9%), with 866 000 new cases and 40 000 deaths in 2018 [1]. Chongqing, a highly densely populated but historically geographically relatively isolated mountainous city, is one of the largest cities in southwest China. Compared with other municipalities, the proportion of rural population is significantly higher, so does its prevalence of TB [3]. Therefore, Chongqing is one of the focuses of TB control in China.
The global distribution of MTBC is characterized by its distinct geographical regionalization [4], and MTB consists of seven lineages, four of which, Indo-Oceanic (lineage 1), East Asian (lineage 2) including W/Beijing family, East African–Indian (lineage 3), and Euro-American (lineage 4) including T family [5,6,7], are the major contributors to TB in humans. Beijing family isolates were reported to be distributed globally and more pathogenic [8], and T family is distributed worldwide as well [7]. More than 80% of the endemic MTB isolates in China belong to Beijing and T families [9, 10].
Several recent studies suggested that the Bayesian population structure analysis method based on the 24-loci mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) could divide each family of MTB into more detailed clades [11,12,13]. The global T family was divided into eight T sub-lineage (TSL1–TSL8) by Bayesian population structure analysis [12], while the Beijing family in China into five Beijing sub-pedigree (BSP1–BSP5) [13]. Interestingly, the clade distribution varies substantially with regions, especially for BSP2 and TSL6, since 99.49% of BSP2 and 89.79% of TSL6 isolates are distributed in Chongqing, compared with that only 0.51% of BSP2 isolates were in Xinjiang, and 4.08, 2.04 and 2.04% of TSL6 in Guizhou, Sichuan, and Jiangsu province, respectively [12, 13].
The observation that a few evolutionary MTBC clades causing TB in humans are geographically restricted [14] has led to our hypothesis that these variants might have adapted to the environment inside local human host [15]. Adaptive evolution refers to the process in which organisms accumulate mutations to adapt to the local environment, resulting in the existence of different populations in their specific regions [16]. Evidence of local adaptation has also been reported in Ghana by two independent studies that lineage 5 of MTB was associated with specific patient ethnicity [16, 17]. Moreover, it was reported that the ancestral populations of MTB in China showed low genetic diversity but with people immigrating, their descendants adapted to the local host environment in different regions, forming the distinct geographical distribution of different evolutionary clades eventually [14, 18]. Accordingly, we are reminded that the distribution and epidemic of BSP2 and TSL6 may be due to local adaptive evolution of the Beijing and T families in Chongqing.
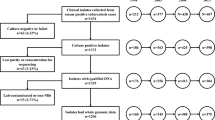
In order to clarify whether BSP2 and TSL6 originated by their local adaptive evolution from the specific clades of Beijing and T families in Chongqing, the genotyping data of 16 090 MTB isolates from laboratory collection in China, published literatures and an open database [19] were collected (Supplementary Table 1). The Bayesian population structure analysis was implemented to mold the consistent clades [12, 13]. The Spacer Oligonucleotide Forest (Spoligoforest) analysis and the 24-loci MIRU-VNTR-based minimum spanning tree (MST) analysis were used to explore the phylogenetic pathways of different families and their clades [20, 21] (Fig. 1). Bayesian demographic history analysis based on the 24-loci MIRU-VNTR data was applied to explore the most possible recent demographic change of clade TSL6 (Fig. 1). The suspected historical events were then considered to speculate the particularity of the host environmental stresses on the local adaptation of the two clades in Chongqing. This study is instructive for deciphering the biogeographic structure and evolutionary history of MTB in Chongqing, China.

The flow chart of data extraction and analysis process. MTB: Mycobacterium tuberculosis; Spoligotyping: Spacer Oligonucleotide typing; MIRU-VNTR: Mycobacterial interspersed repetitive unit-variable number of tandem repeats; Spoligoforest: Spacer Oligonucleotide Forest; TSL: T sub-lineage; BSP: Beijing sub-pedigree
Methods
Data collection
The study was based on genotyping and geographical distribution data of a collection of MTB isolates (n = 16 090) from China. These data were mainly collected from the published literatures (n = 15 830), and there were also a small collection from a key laboratory of Sichuan University (n = 193) [13] and the SITVIT2 database (http://www.pasteur-guadeloupe.fr:8081/SITVIT) (n = 67), respectively [19] (Supplementary Table 1). In the collected data, spoligotype international types (SIT) information of 12 674 isolates was available and listed (Supplementary Table 2), and the 24-loci MIRU-VNTR data of 2161 isolates amongst all the data collected were available (Supplementary Table 3).
Bayesian population structure analyses
Bayesian population structure analyses were conducted on the 1742 isolates of Beijing family and T family amongst the above isolations with 24-loci MIRU-VNTR data (Supplementary Table 4). The STRUCTURE software (version 2.3) (Pritchard Lab, Stanford University, Stanford, CA, USA) [22] was used by implementing an admixture model considering that the data originated from the admixture of K ancestral populations at some time in the past. Posterior estimates for the parameters of interest were computed by using a Markov chain Monte Carlo (MCMC) algorithm in ten parallel chains with a burn-in of 100 000 iterations and a run length of 106. The delta K was calculated using the program STRUCTURE HARVESTER by the Evanno method [23]. Medians were calculated from 10 replicates for K by using the FullSearch algorithm implemented in CLUMPP 1.1.2 software (Stanford University, Stanford, CA, USA) to guarantee the optimum clustering [24], and a cutoff of 0.6 was fixed for clustering of isolates.
Phylogenetic inferences
The drawing of Spoligoforest (Fig. 2) used the spolTools software (http://www.emi.unsw.edu.au/spolTools) and the drawing of MST based on data of 24-loci MIRU-VNTR (Fig. 3) used BioNumerics software 6.6 (Applied Maths, Sint-Martens-Latem, Belgium). The identical MIRU-VNTR haplotypes in the MST were pooled as a single node representing a cluster, and the rate of clustered isolates was considered as an indicator for the extent of recent transmission.

Spoligoforest tree illustrating evolution of MTB isolates in China (n = 12 674). Different color represents different families; Numbers outside parentheses represent the matched SIT ID in the Pasteur database; The numbers in parentheses are the quantity of isolates; Probable strain phylogenetic pathways between spoligotypes, solid black lines represent links of weigh being 1.0, dashed lines represent links of weight between 0.5 to 1, dotted lines represent links of weight less than 0.5. MTB: Mycobacterium tuberculosis; SIT: Spoligotype international type; CAS: Central-Asian family; H: Haarlem family; LAM: Latin American and Mediterranean family; EAI: East-African Indian family

MSTs illustrating the phylogenetic pathways. The MST connects each genotype based on degree of changes required to go from one allele to another; The lines denotes the number of allele changes between two patterns: less than three changes (solid lines), four changes (gray dashed lines) and five or more changes (gray dotted lines); The size of the circle is proportional to the quantities of isolates sharing same pattern. MST: Minimum Spanning Tree; CAS: Central-Asian family; H: Haarlem family; LAM: Latin American and Mediterranean family; EAI: East-African Indian family; TSL: T sub-lineage; BSP: Beijing sub-pedigree
WebLogo of allele
WebLogo (http://weblogo.berkeley.edu/logo.cgi) [25] was used to visualize main patterns of tandem repeats for 24-loci MIRU-VNTRs and showed copy number of 24 MIRU-VNTR loci in MTB clades BSP2, BSP4, TSL5 and TSL6.
Demographic history analysis of TSL6
The Bayesian approach [26] that assumed a stepwise mutation model (SMM) [27, 28] was used to estimate the posterior distribution of demographic and genealogical parameters [11]. The parameters of the clade TSL6 (n = 48) was estimated by using 24-loci MIRU-VNTRs with MCMC simulations implemented in the Msvar 1.3 algorithm. The assumed demographic history is based on a past population of size N1 that experienced a change in size at some time ta in the past to reach current effective population size N0. We tested hypothesis of declining population (10− 2 and 10− 3 as a prior) where expansion ratio R < 1 (R = N0/N1), of stable population where R = 1 and of expanding populations (101 to 103 as a prior) where R > 1. The analyses were performed assuming exponential demographic change. The prior mutation rate value of each MIRU-VNTR locus ranged between 1.55 × 10− 8 and 6.65 × 10− 8 per locus and per generation, as claimed by previous studies [11, 20, 29]. The chain was run for 1.2 billion steps, with parameter values recorded every 100 000 steps. The MCMC output was analyzed using the software Tracer (GISUM group, University of Málaga, Málaga, Spain) [30] to obtain the posterior distribution and the effective sample size (ESS) of all parameters (which were all above 168) after a burn-in of 10%.
Results
Spoligoforest analysis
The evolution and genetic distance of the different MTB families were demonstrated by the Spoligoforest (Fig. 2) created with the available Spoligotyping data of the collected isolates (n = 12 674). The Spoligoforest tree showed that Beijing family and T family composed two of the most predominant families of the isolates collected. The collected Beijing family isolates showed a low diversity of SIT, and the main Beijing family isolates belong to SIT1 (n = 9121, 94.37%). The collected T family isolates were more diverse than Beijing family on SIT, consisting of SIT53 (n = 689, 40.67%), SIT52 (n = 267, 15.76%), SIT334 (n = 90, 5.31%), SIT37 (n = 74, 4.37%), etc. The Spoligoforest tree showed that Beijing family and T family appeared as different aggregates.
The Bayesian population structure
Implementing of the Bayesian population structure analyses divided the collected isolates of Beijing family that with available 24-loci MIRU-VNTR data into six STRUCTURE clades (n = 1490), while the collected T isolates belong to six STRUCTURE clades (n = 252). These STRUCTURE clades were defined and labeled in accordance with previous researches (BSP1–5, BSPint and TSL3–6, TSL8, TSLint) (Fig. 3b, Supplementary Table 4). Note that BSPint and TSLint to represent isolates in intermediate position among various clades defined in evolution.
MST analysis based on 24-loci MIRU-VNTR
The MST reconstructed from the pooled data for available isolates with 24-loci MIRU-VNTR analysis data (n = 2161) highlighted the phylogenetic pathway between the different isolates mentioned previously (Fig. 3a), including Beijing family (n = 1767), T family (n = 252), Manu family (n = 302), Central-Asian family (CAS) (n = 18), Haarlem family (H) (n = 16), Latin American and Mediterranean family (LAM) (n = 10), East-African Indian family (EAI) (n = 1) and others (n = 49). Despite that they are genetically far apart, yet obvious intersections and overlaps between the Beijing family and the T family in the MST were observed. The MST of available isolates of different STUCTURE clades (n = 1742) indicated that BSP2 and TSL6 are directly downstream of the phylogenetic pathway of BSP4 and TSL5, respectively (Fig. 3b). Moreover, the clades BSP3, BSP4, and most of the BSP5, displayed a star-like shape of phylogenetic expansion in MST while BSP2 and TSL6 displayed a polycentric shape. In addition, the MST analysis and the WebLogo of allele copy number both showed that BSP2 and TSL6 had the similar main patterns of 24-loci MIRU-VNTR (Figs. 3b, 4, Supplementary Tables 4 and 5).

WebLogo of allele copy of 24 MIRU-VNTRs loci of clades BSP2, BSP4, TSL5, and TSL6. MIRU-VNTR: Mycobacterial interspersed repetitive unit-variable number of tandem repeats; TSL: T sub-lineage; BSP: Beijing sub-pedigree
The recent demographic change of TSL6
The SMM in the Msvar 1.3 algorithm deduced the most probable past demographic history of TSL6 in China (Fig. 5). In the result, ta = 2.0463 (log scale), which computed that the past demographic history of TSL6 clade in China might have taken 111.25 years while the mutation rate per locus and per generation was 7.24 × 10− 7.

Demographic analysis and dating estimates of clade TSL6. ta, time elapsed since last expansion in years (log scale); R = N0/N1, median value of expansion ratio; μ, mutation rate per locus and per generation. All estimates correspond to median values, followed by 95% highest posterior densities indicated in parentheses. TSL: T sub-lineage
Discussion
The logical local adaptive evolution of BSP2 and TSL6 in Chongqing
In this study, our phylogenetic analysis suggested the clades BSP2 and TSL6 might evolve from BSP4 and TSL5, respectively, and this was consistent with the recent studies by Reynaud and Zheng [12, 13]. However, according to the geographical distribution (Supplementary Table 4) and the results of the recent study by Reynaud and Zheng, the main epidemic region of TSL5 was in Jiangsu province [12], while BSP4 in the mainland of China was epidemic in Tibet [13]. Therefore, BSP2 and TSL6 might be caused by the genetic polymorphisms generated after the entry of extraneous isolates into Chongqing from Tibet and Jiangsu.
It was observed that the phylogeny of BSP2 and TSL6 isolates appeared in the polycentric shape in MST, indicating that the evolutionary expansions of BSP2 and TSL6 were drastic [20] (Fig. 3b). Considering that the expansion and evolution of MTB were mainly caused by human activities such as migration [18, 20, 28], the emerging of BSP2 and TSL6 might result from a drastic historical event. Our Bayesian demography analysis showed that stepwise mutation of TSL6 has lasted about 111 years (Fig. 5), which was likely to be started from the 1900s. Back to Chinese history, as a result of the forced sign of the “Treaty of Shimonoseki” in 1895, five trading ports were opened up [31]. Suzhou in Jiangsu province became the trading port due to its sea location while Chongqing became one of the first batches of inland commerce ports opened to foreigners. The British, French, German, American and Japanese consulates were stationed in Chongqing during 1890–1904, and many densely populated industries sprung up in Chongqing during this period. Frequent commercial traffic along the trade routes at that time could likely carry TSL5 isolates from Jiangsu to Chongqing, where TSL5 might survive and present stably because it was reported that T family could coexist well with other families [14]. The Tea-horse Ancient Road, which connected southwest to west China from the Tang dynasty to the early 20 century [32], might make great contribution to the appearance of BSP4 in Chongqing at a much earlier time than TSL5 did, leading to BSP4 becoming an aboriginal clade in this area as in most other regions in China, since a few upstream in the phylogenetic pathway of MTB clades in southwestern and central China came from Tibet [33]. We would speculate that BSP4 might have undergone a certain degree of local adaptive evolution in Chongqing before the entrance of TSL5, therefore only demographic analysis for TSL6 was performed in this study. Besides, the success of MTB as a pathogen was largely depend on the host adaptation and selection [14], so the arrival of the Europeans and the emergence of densely populated industries might exert an impact on the original host environment and thus created a new hosting type. Hence the isolates of TSL5 and BSP4 might have to start to adapt to this new host environment actively. Moreover, the research of Liu et al. proved that the various clades of MTBC in China evolved from the ancestors with very low genetic diversity [18] so that distinct genetic polymorphisms of clades of MTB might result from local adaptive evolution [14]. Giving the above evidence, BSP2 and TSL6 clades could be originated from BSP4 and TSL5 by their local adaptive evolution in Chongqing.
The convergence of the pattern at MIRU-VNTR of BSP2 and TSL6
A distinct feature of BSP2 and TSL6 isolates was the similar pattern of 24-loci MIRU-VNTR (Fig. 4). Our result indicated that some isolates from the BSP4 and TSL5 clades evolved to give a similar 24-loci MIRU-VNTR pattern during their local adaptive evolution in Chongqing, initiating the formation of BSP2 and TSL6 (Fig. 3). There are clear differences between the 24-loci MIRU-VNTR repeat patterns of BSP4 and T5, which of BSP2 and TSL6, however, are similar in all loci (Fig. 4). Local adaptive evolution is a common phenomenon among MTB, and the host environment of MTB in different regions are complex and diverse [14]. The convergence of MIRU-VNTR might have satisfied the need of the isolates to adapt to the local host environment. Like most of satellite DNA, MIRU-VNTR loci can affect some traits via regulating the expression of upstream and downstream genes or others through quantitative changes of repeats [34,35,36,37,38,39,40]. For example, the repeat number of QUB26 could directly affect the expression of Rv3610, thus its over-expressed product, FtSH, a membrane protein of MTB, could significantly affect the growth and viability of MTB [35,36,37]. Although the changes in the number of repeats at the MIRU-VNTR loci in local adaptive evolution and their effect on MTB traits remain to be further studied, it is reasonable to propose that the convergence of 24-loci MIRU-VNTR repeat patterns may indeed lead to the similarity of the growth and virulence traits of MTB isolates. Filliol et al. confirmed that MIRU-VNTR were based on a limited number of loci, and the markers used evolved rapidly with a tendency to converge [41], therefore the convergence of MIRU-VNTR patterns might be one manifestation of the local adaptive evolution of BSP2 and TSL6, which may also be true in other clades.
The limitations of 24-loci MIRU-VNTR in discriminatory power
Our results showed that similar MIRU-VNTR pattern could occur in the different clades of Beijing and T families in Chongqing (Figs. 3, 4, Supplementary Table 5), although Spoligotyping showed that they were genetically distant (Fig. 2, Supplementary Table 4), indicating the insufficient discriminatory power of 24-loci MIRU-VNTR on genotyping of specific clades of Beijing and T families. Murase et al. exhibited that the discriminatory power of particular MIRU-VNTR loci varied depending on the specific strain background [42]. Moreover, the properties of molecular markers required to address at both local and global levels of bacterial diversity are unlikely to be met by one single marker, hence, the 24-loci MIRU-VNTR should be combined with robust markers for comprehensive genotyping, especially for some clades like BSP2 and TSL6. The robust single nucleotide polymorphisms (SNP) markers can be used to construct high resolution and reproducible phylogenies [5]. Spoligotyping and genotyping methods based on large sequence polymorphisms (LSP) have been started to be applied on the classification of MTB in various lineages [43], and the core genome multi-locus sequence typing (cgMLST) based Whole Genome Sequencing analyses facilitate the discrimination of longitudinal MTBC outbreaks of high resolution [44]. From what have been known, the combination of 24-loci MIRU-VNTR and the robust makers can lead to the potent and universal comprehensive genotyping approach, and MIRU-VNTR loci should be used in a lineage-dependent manner for epidemiological purposes.
Conclusion
By the analyses of Bayesian population structure, phylogenetic pathway, and demographic history, it was revealed that BSP2 and TSL6 clades could be originated from BSP4 and TSL5 by their local adaptive evolution in Chongqing, although this may need further verification by applying whole genome sequencing to accumulate its robustness. Our results suggest MIRU-VNTR be combined with other robust markers for a more comprehensive genotyping approach, especially for families with clades of the same MIRU-VNTR pattern.
Availability of data and materials
Data of Spoligotyping and data of MIRU-VNTR generated and analyzed in this manuscript were listed in the supplementary files and part of them were deposited in a public database, SITVIT2 (http://www.pasteur-guadeloupe.fr:8081/SITVIT2/).
Abbreviations
- MTB:
-
Mycobacterium tuberculosis
- MIRU-VNTR:
-
Mycobacterial interspersed repetitive unit-variable number of tandem repeats
- Spoligoforest:
-
Spacer Oligonucleotide Forest
- MST:
-
Minimum spanning tree
- MTBC:
-
M. tuberculosis complex
- TSL:
-
T sub-lineage
- BSP:
-
Beijing sub-pedigree
- Spoligotyping:
-
Spacer Oligonucleotide typing
- MCMC:
-
Markov chain Monte Carlo
- SMM:
-
Stepwise mutation model
- ESS:
-
Effective sample size
- SIT:
-
Spoligotype international type
- CAS:
-
Central-Asian family
- H:
-
Haarlem family
- LAM:
-
Latin American and Mediterranean family
- EAI:
-
East-African Indian family
- SNP:
-
Single nucleotide polymorphisms
- LSP:
-
Large sequence polymorphisms
- cgMLST:
-
Core genome multi-locus sequence typing
References
World Health Organization. Global tuberculosis report 2019. 2019.
Braveman P, Gruskin S. Poverty, equity, human rights and health. Bull World Health Organ. 2003;81(7):539–45.
Zhang D, An J, Wang J, Hu C, Wang Z, Zhang R, et al. Molecular typing and drug susceptibility of Mycobacterium tuberculosis isolates from Chongqing municipality, China. Infect Genet Evol. 2013;13:310–6.
Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26(6):431–44.
Coll F, McNerney R, Guerra-Assuncao JA, Glynn JR, Perdigao J, Viveiros M, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812.
Tessema B, Beer J, Merker M, Emmrich F, Sack U, Rodloff AC, et al. Molecular epidemiology and transmission dynamics of Mycobacterium tuberculosis in Northwest Ethiopia: new phylogenetic lineages found in Northwest Ethiopia. BMC Infect Dis. 2013;13:131.
Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, et al. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23.
Ribeiro SC, Gomes LL, Amaral EP, Andrade MR, Almeida FM, Rezende AL, et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol. 2014;52(7):2615–24.
Lu W, Lu B, Liu Q, Dong H, Shao Y, Jiang Y, et al. Genotypes of Mycobacterium tuberculosis isolates in rural China: using MIRU-VNTR and spoligotyping methods. Scand J Infect Dis. 2014;46(2):98–106.
Dong H, Liu Z, Lv B, Zhang Y, Liu J, Zhao X, et al. Spoligotypes of Mycobacterium tuberculosis from different provinces of China. J Clin Microbiol. 2010;48(11):4102–6.
Reynaud Y, Millet J, Rastogi N. Genetic structuration, demography and evolutionary history of Mycobacterium tuberculosis LAM9 sublineage in the Americas as two distinct subpopulations revealed by Bayesian analyses. PLoS One. 2015;10(10):e0140911.
Reynaud Y, Zheng C, Wu G, Sun Q, Rastogi N. Bayesian population structure analysis reveals presence of phylogeographically specific sublineages within previously ill-defined T group of Mycobacterium tuberculosis. PLoS One. 2017;12(2):e0171584.
Zheng C, Reynaud Y, Zhao C, Zozio T, Li S, Luo D, et al. New Mycobacterium tuberculosis Beijing clonal complexes in China revealed by phylogenetic and Bayesian population structure analyses of 24-loci MIRU-VNTRs. Sci Rep. 2017;7(1):6065.
Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 2018;16(4):202–13.
Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond Ser B Biol Sci. 2012;367(1590):850–9.
Kawecki T, Ebert D. Conceptual issues in local adaptation. Ecol Lett. 2004;7:1225.
Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12(8):581–91.
Liu Q, Ma A, Wei L, Pang Y, Wu B, Luo T, et al. China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat Ecol Evol. 2018;2(12):1982–92.
Couvin D, Rastogi N. Tuberculosis - a global emergency: tools and methods to monitor, understand, and control the epidemic with specific example of the Beijing lineage. Tuberculosis (Edinb). 2015;95(Suppl 1):S177–89.
Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 2015;47(3):242–9.
Chatterjee A, Mistry NJT. MIRU–VNTR profiles of three major Mycobacterium tuberculosis spoligotypes found in western India. Tuberculosis. 2013;93(2):250–6.
Earl DA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359–61.
Evanno GS, Regnaut SJ, Goudet JJME. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–20.
Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23(14):1801–6.
Olsen LR, Kudahl UJ, Simon C, Sun J, Schonbach C, Reinherz EL, et al. BlockLogo: visualization of peptide and sequence motif conservation. J Immunol Methods. 2013;400–401:37–44.
Storz JF, Beaumont MA. Testing for genetic evidence of population expansion and contraction: an empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution. 2002;56(1):154–66.
Wirth T, Hildebrand F, Allix-Beguec C, Wolbeling F, Kubica T, Kremer K, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 2008;4(9):e1000160.
Barbier M, Wirth T. The evolutionary history, demography, and spread of the Mycobacterium tuberculosis complex. Microbiol Spectr. 2016;4(4). https://doi.org/10.1128/microbiolspec.TBTB2-0008-2016..
Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45(7):784–90.
Longair MH, Baker DA, Armstrong JD. Simple Neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics. 2011;27(17):2453–4.
Miller FP, Vandome AF, McBrewster J, Shimonoseki, Yamaguchi, Qing. Treaty of Shimonoseki. Washington: Alphascript Publishing; 2010.
Freeman M, Ahmed S. Tea horse road: China’s ancient trade road to Tibet. 1st ed. Bangkok: River Books Press; 2011.
Yin QQ, Liu HC, Jiao WW, Li QJ, Han R, Tian JL, et al. Evolutionary history and ongoing transmission of phylogenetic sublineages of Mycobacterium tuberculosis Beijing genotype in China. Sci Rep. 2016;6:34353.
Tantivitayakul P, Panapruksachat S, Billamas P, Palittapongarnpim PJT. Variable number of tandem repeat sequences act as regulatory elements in Mycobacterium tuberculosis. Tuberculosis. 2010;90(5):311–8.
Kiran M, Chauhan A, Dziedzic R, Maloney E, Rajagopalan MJT. Mycobacterium tuberculosis ftsH expression in response to stress and viability. Tuberculosis. 2009;89(Suppl 1):S70–3.
Ito K, Akiyama Y. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol. 2005;59(1):211–31.
Pérez-Lago L, Navarro Y, Herranz M, Bouza E, García-de-Viedma D. Differences in gene expression between clonal variants of Mycobacterium tuberculosis emerging as a result of microevolution. Int J Med Microbiol. 2013;303(8):674–7.
Akhtar P, Singh S, Bifani P, Kaur S, Srivastava BS, Srivastava R. Variable-number tandem repeat 3690 polymorphism in Indian clinical isolates of Mycobacterium tuberculosis and its influence on transcription. J Med Microbiol. 2009;58(6):798–805.
Zhou AP, Xu ZH, Sun Q, Yao YF, Wu XF. Research on relationship between polymorphisms of Mtub-39 and expression of downstream genes of Mycobacterium tuberculosis. J Shanghai Jiaotong Univ. 2012;32(11):1415–20 (in Chinese).
Smittipat N, Palittapongarnpim P. Identification of possible loci of variable number of tandem repeats in Mycobacterium tuberculosis. Tuber Lung Dis. 2000;80(2):69–74.
Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, et al. Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 2006;188(2):759–72.
Murase Y, Mitarai S, Sugawara I, Kato S, Maeda S. Promising loci of variable numbers of tandem repeats for typing Beijing family Mycobacterium tuberculosis. J Med Microbiol. 2008;57(Pt 7):873–80.
Ei PW, Aung WW, Lee JS, Choi G-E, Chang CL. Molecular strain typing of Mycobacterium tuberculosis: a review of frequently used methods. J Korean Med Sci. 2016;31(11):1673–83.
Kohl TA, Diel R, Harmsen D, Rothganger J, Walter KM, Merker M, et al. Whole-genome-based Mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach. J Clin Microbiol. 2014;52(7):2479–86.
Acknowledgements
Not applicable.
Funding
This work was financially supported by the Department of Science and Technology of Sichuan (18GJHZ0137). This funding source had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Peng-Kuan Liang and Chao Zheng contributed to the work equally and should be regarded as co-first authors. Peng-Kuan Liang, Chao Zheng and Qun Sun conceived the study; Chao Zheng, Chang-Song Zhao, Peng-Kuan Liang, Yann Reynaud, Thierry Zozio, David Couvin and Nalin Rastogi collected the data; Peng-Kuan Liang, Chao Zheng, David Couvin, Yann Reynaud and Thierry Zozio analyzed the data; Peng-Kuan Liang, Xiao-Fang Xu and Zhe-Ze Zhao wrote the manuscript; Yann Reynaud and Chang-He Li read and approved the manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary information
Additional file 1
: Table S1. The source of available data in this study.
Additional file 2
: Table S2. The Spoligotype International Type (SIT) information.
Additional file 3
: Table S3. The 24-loci MIRU-VNTR data of 2161 isolates for MST building.
Additional file 4
: Table S4. Dataset used in the MST of T and Beijing families for the 1742 MTB isolates.
Additional file 5
: Table S5. Repeat number of 24-loci MIRU-VNTR loci in MTB isolates of clades BSP2 and TSL6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, PK., Zheng, C., Xu, XF. et al. Local adaptive evolution of two distinct clades of Beijing and T families of Mycobacterium tuberculosis in Chongqing: a Bayesian population structure and phylogenetic study. Infect Dis Poverty 9, 59 (2020). https://doi.org/10.1186/s40249-020-00674-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40249-020-00674-7