
Abstract
Background
The LDL-C lowering effect of ezetimibe has been attributed primarily to increased catabolism of LDL-C via up-regulation of LDL receptor (LDLR) and decreased cholesterol absorption. Recently, ezetimibe has been demonstrated to have reverse cholesterol transport (RCT) promoting effects in mice, hamsters and humans. However, the underlying mechanisms are still not clear. The aim of this study is to investigate whether ezetimibe improves RCT-related protein expression in LDLR−/− hamsters.
Methods
A high-fat diet was used to induce a human-like hyperlipidemia in LDLR−/− hamsters. Lipid profiles were assayed by commercially available kits, and the effects of ezetimibe on lipid metabolism-related protein expression were carried out via western blot.
Results
Our data demonstrated that ezetimibe administration significantly reduced plasma total cholesterol (~ 51.6% reduction, P < 0.01) and triglyceride (from ~ 884.1 mg/dL to ~ 277.3 mg/dL) levels in LDLR−/− hamsters fed a high-fat diet. Ezetimibe administration (25 mg/kg/d) significantly promoted the protein expression of cholesterol 7 alpha-hydroxylase A1 (CYP7A1), LXRβ and peroxisome proliferator-activated receptor (PPAR) γ; and down-regulated the protein expression of PPARα and PPARβ. However, it showed no significant effect on sterol regulatory element-binding protein (SREBP)-1c, SREBP-2, proprotein convertase subtilisin/kexin type 9 (PCSK9), Niemann-Pick C1-like 1 (NPC1L1), and ATP-biding cassette (ABC) G5/G8.
Conclusion
Ezetimibe may accelerate the transformation from cholesterol to bile acid via promoting CYP7A1 and thereby enhance RCT. As a compensatory mechanism of TG lowering, ezetimibe promoted the protein expression of PPARγ and decreased PPARα and β. These results are helpful in explaining the lipid-lowering effects of ezetimibe and the potential compensatory mechanisms.
Similar content being viewed by others


Background
Hyperlipidemia is an important pathogenic factor of atherosclerosis, which is the basic pathological change of cardiovascular disease [1]. On average, ~ 50% of cholesterol in the gut lumen is reabsorbed in humans and rodents [2, 3], and the remainder excreted in feces. Decreased intestinal absorption of cholesterol is associated with reduced mortality from atherosclerotic cardiovascular disease. Therefore, inhibition of intestinal absorption of cholesterol is a good way to reduce hyperlipidemia and atherosclerotic cardiovascular disease.
Ezetimibe is developed as a cholesterol absorption inhibitor mainly via targeting Niemann-Pick C1-like 1 (NPC1L1) in the intestine [4]. In humans, once-daily treatment with ezetimibe monotherapy (10 mg) inhibits cholesterol absorption on average by 54–65% and results in a ~ 20% reduction of plasma low-density lipoprotein (LDL) cholesterol (LDL-C) [5]. In high-fat fed rhesus monkeys, ezetimibe causes ~ 69% of cholesterol reduction in chylomicrons during the postprandial phase which indirectly lead to a decrease of LDL-C and particle number [6]. It is worthy to note that recent studies suggested that ezetimibe may also promote reverse cholesterol transport (RCT) [7, 8], which plays a key role in cholesterol homeostasis.
RCT is a multi-step process that involves the efflux of cholesterol from peripheral cells to plasma, the hepatic uptake, the biliary excretion, and fecal excretion [9]. Biliary cholesterol can be derived from at least three distinct sources: hepatic de novo cholesterol synthesis, hepatic cholesterol stores, or cholesterol cleared from plasma lipoproteins [10]. The transintestinal cholesterol excretion (TICE) route, independent on biliary route in RCT, for cholesterol excretion in the feces was also provided [11]. Tang et al. viewed all these pathways as a cholesterol transport system [12].
A previous report demonstrated that treatment of C57BL/6 J mice with ezetimibe decreased dietary cholesterol absorption by 86% and increased RCT from peripheral tissue macrophages by 6-fold [13]. Xie et al. suggested that ezetimibe can promote macrophage RCT via inhibition of hepatic NPC1L1 function in transgenic liver-only human NPC1L1 mice [3]. Altemus et al. revealed that the macrophage-to-feces RCT promoting effect of ezetimibe was associated with increased expression of hepatic ABCG5 and ABCG8, which was due to the inhibition of intestinal cholesterol absorption [14]. However, Wang et al. demonstrated that ezetimibe may promote cholesterol elimination via an ABCG5/G8-independent pathway [15]. In hyperlipidemic patients, ezetimibe treatment approximately doubled the flux of plasma-derived cholesterol into fecal neutral sterols, in association with increases in fractional clearance rate of plasma cholesterol ester, and plasma de novo cholesterol synthesis [10]. These studies indicated that ezetimibe may promote the net transfer of cholesterol from peripheral cells for ultimate elimination in the feces. However, some studies have challenged the hypothesis that ezetimibe increases RCT through decreased reabsorption of biliary cholesterol in the intestine [16].
One of the limitations of the previous studies in mice is that mice are high-density lipoprotein cholesterol (HDL-C) dominant, while humans are non-HDL-C dominant [17]. Furthermore, mice are naturally lack cholesteryl ester transfer protein, which is important for cholesterol metabolism. Therefore, the results collected from mice may not reflect the actual situations in humans. Furthermore, the previous results in mice seem to be inconsistent between groups, and the underlying mechanisms need to be further confirmed by other animal models [14, 15]. Although the studies in humans have elucidated some useful phenomena, the underlying mechanisms are not convenient to be carried out in humans. Therefore, it is interesting to study whether ezetimibe improves RCT by improving RCT-related protein expression in animals whose lipid profile is close to that of humans.
Golden Syrian hamster is known to develop human-like hyperlipidemia following a high-fat diet [18]. Additionally, the LDL-C lowering effect of ezetimibe is usually attributed primarily to increased catabolism of LDL-C via up-regulation of LDL receptor (LDLR) [19]. Therefore, it is interesting to investigate whether ezetimibe improves RCT-related protein expression in LDLR−/− hamsters fed a high-fat diet, and other potential effects beyond what is presently known.
Methods
Materials
Ezetimibe was the product of Selleck (Shanghai, China). High-fat diet (21% fat and 0.25% cholesterol) was provided by Beijing HFK Bioscience Co., Ltd. Complete protease inhibitor cocktail tablets were purchased from Roche (Schweiz, Germany). RIPA lysis buffer was a product of Solarbio (Beijing, China). Rabbit polyclonal antibody against Liver X receptor (α) and LXRβ, and rabbit monoclonal antibody against scavenger receptor B type 1 (SR-B1) and LDLR were from Abcam (Cambridge, MA, USA). Mouse monoclonal antibody against peroxisome proliferator-activated receptor α (PPARα), PPARβ and PPARγ, and cholesterol 7 alpha-hydroxylase A1 (CYP7A1), Niemann-Pick C1-like 1 (NPC1L1), sterol regulatory element-binding protein (SREBP)-1c and SREBP-2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse monoclonal antibody against β-actin and rabbit monoclonal antibody against proprotein convertase subtilisin/kexin type 9 (PCSK9), and rabbit polyclonal antibody against ATP-biding cassette (ABC) G5 were the products of Proteintech (Chicago, IL, USA). Mouse monoclonal antibody against ABCG8 and enhanced chemiluminescence (ECL) kits were purchased from Thermo Scientific Pierce (Rockford, IL, USA). All reagents used in this study were of analytical grade.
Animals and grouping
Ten LDLR−/− Golden Syrian hamsters (male, 165 ± 15 g) were provided by prof. George Liu at Peking University (Beijing, China). All experiments were approved by the Laboratory Animal Ethical Committee of Weifang Medical University and followed the NIH guidelines for the care and use of animals. LDLR−/− hamsters were fed a high-fat diet. After a one-week adaptive period, the hamsters were randomly divided into two groups, the model group (0.9% sodium chloride by gavage, n = 5), and the ezetimibe group (25 mg/kg/d by gavage, n = 5). The hamsters were treated for 4 weeks, then they were sampled.
Plasma analysis
Total cholesterol (TC) and triglyceride (TG) levels in the plasma were determined enzymatically using commercially available assay kits of Biosino Biotechnology and Science, Inc. (Beijing, China) according to the instructions. The mixed plasma of each group was further separated by fast protein liquid chromatography (FPLC). Briefly, 150 μL plasma was loaded onto a Superose™ 6 10/300 gel chromatography column connected to ÄKTA-FPLC system, and eluted with normal saline at a flow rate of 0.3 mL/min [20]. Eluted fractions (0.5 mL per tube) were collected, and the lipid content was assayed using the same TC and TG kits.
Protein isolation, electrophoresis, and western blotting
Total proteins from the liver and small intestine were extracted using RIPA lysis buffer with complete protease inhibitor according to the manufacturer’s instructions. Equal amounts of protein (20 μg) were subjected to 6% or 10% SDS-PAGE and transferred onto Polyvinylidene fluoride membranes by electroblotting. After blocking in Tris-buffered saline containing 0.1% Tween-20 and 5% nonfat dry milk for 2 h at room temperature, the membranes were incubated with primary antibodies overnight at 4 °C. After washing 3 times, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. Immunoblots were revealed by enhanced chemiluminescence reaction and images were captured by Clinx ChemiScope 6000 Pro (Shanghai, China). Densitometry analysis was conducted using Image-Pro Plus software version 6.0 (Media Cybernetics Corp, Bethesda, MD, USA) and normalized by housekeeping protein β-actin [21, 22].
Data analysis
All of the bioassay results were expressed as the mean ± standard deviation (SD) for at least three independent experiments. Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Tukey’s test. Differences were considered to be significant at a P < 0.05.
Results
Ezetimibe lowered plasma TC and TG levels in LDLR−/− hamsters
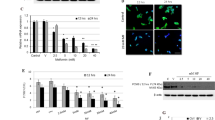
As shown in Fig. 1a, ezetimibe administration significantly reduced plasma TC of the LDLR−/− hamsters fed a high-fat diet when compared with the model group (~ 51.6% reduction, P < 0.01). Furthermore, ezetimibe treatment lowered the TG levels of the LDLR−/− hamsters from ~ 884.1 mg/dL to ~ 277.3 mg/dL (Fig. 1b, ~ 68.6% reduction, P < 0.01). Further lipid profile analysis after ÄKTA-FPLC separation indicated that ezetimibe treatment significantly decreased the cholesterol levels of very low-density lipoprotein (VLDL) and LDL particles, and exhibited comparably lower effect on HDL-C (Fig. 1c). Furthermore, ezetimibe administration also significantly lowered the TG content of VLDL, LDL, and HDL particles (Fig. 1d).

Effect of ezetimibe on the plasma profile of the LDLR−/− hamsters fed a high-fat diet (n = 5). a, ezetimibe lowers plasma TC of the LDLR−/− hamsters; b, ezetimibe lowers plasma TG of the LDLR−/− hamsters; c, TC profiles in different lipoprotein fractions after ÄKTA-FPLC separation; d, TG profiles in different lipoprotein fractions after ÄKTA-FPLC separation. Data are expressed as mean ± SD. **p < 0.01 vs model group
Ezetimibe improved CYP7A1 expression in the liver of the LDLR−/− hamsters
In this study, ezetimibe treatment showed no significant effect on the protein expression of SR-B1 (Fig. 2a), which plays a key role in hepatic uptake of HDL-C [9, 12]. LDLR delivers non-HDL particles to the liver, and PCSK9 binds LDLR and leads to its degradation in the endosome [23]. In LDLR−/− hamsters, the protein expression of LDLR was not detectable (data not shown), and ezetimibe administration exhibited no significant effect on the protein expression of PCSK9 in this study (Fig. 2b). CYP7A1 is the first rate-limiting enzyme of bile acid synthesis. It is worthy to note that ezetimibe treatment significantly promoted the protein expression of CYP7A1 compared to the model group (~ 2.1-fold, P < 0.01, Fig. 2c). However, ezetimibe treatment showed no significant effect on the protein expression of ABCG5 and ABCG8 (Fig. 2d and e), which play key roles in the transport of bile acid and cholesterol to the gall bladder for excretion.

Effect of ezetimibe on the RCT-related protein expression in the liver of the LDLR−/− hamsters (n = 3). a, protein expression of SR-B1 and densitometric quantification; b, protein expression of PCSK9 and densitometric quantification; c, protein expression of CYP7A1 and densitometric quantification; d, protein expression of ABCG5 and densitometric quantification; e, protein expression of ABCG8 and densitometric quantification. Data are expressed as mean ± SD. **p < 0.01 vs model group
Ezetimibe down-regulated PPARα and β and up-regulated PPARγ expression in the liver of the LDLR−/− hamsters
SREBPs are important transcription factors involved in the regulation of lipid metabolism and homeostasis. In the present study, ezetimibe showed no significant influence on the protein expression of both SREBP-2 and SREBP-1c (Fig. 3a and b). PPARs are key regulators of lipid storage, especially TG metabolism and utilization of fat. In contrast to the model group, ezetimibe treatment significantly reduced the protein expression of both PPARα and β (~ 32.5%, P < 0.05, Fig. 3c and d). Furthermore, ezetimibe administration significantly promoted the protein expression of PPARγ compared with the model group (~ 1.5-fold, P < 0.05, Fig. 3e).

Effect of ezetimibe on the protein expression of SREBPs and PPARs in the liver of the LDLR−/− hamsters (n = 3). a, protein expression of SREBP-2 and densitometric quantification; b, protein expression of SREBP-1c and densitometric quantification; c, protein expression of PPARα and densitometric quantification; d, protein expression of PPARβ and densitometric quantification; e, protein expression of PPARγ and densitometric quantification. *p < 0.05 vs model group
Ezetimibe up-regulated LXRβ expression in the small intestine of the LDLR−/− hamsters
LXRs regulate genes that encode proteins involved in lipid transport and excretion. In this study, ezetimibe showed no significant effect on the protein expression of LXRα (Fig. 4a). It is worthy to note that ezetimibe dramatically promoted the protein expression of LXRβ when compared to the model group (~ 1.8-fold, P < 0.01, Fig. 4b). However, ezetimibe treatment showed no significant effect on the protein expression of ABCG5 and ABCG8 (Fig. 4c and d). Furthermore, ezetimibe showed no effect on the protein expression of NPC1L1 compared with the model group (Fig. 4e). This result further suggested that ezetimibe inhibits the function rather than the protein expression of NPC1L1.

Effect of ezetimibe on the protein expression of LXR/ABC transporters and NPC1L1 in the small intestine of the LDLR−/− hamsters (n = 3). a, protein expression of LXRα and densitometric quantification; b, protein expression of LXRβ and densitometric quantification; c, protein expression of ABCG5 and densitometric quantification; d, protein expression of ABCG8 and densitometric quantification; e, protein expression of NPC1L1 and densitometric quantification. **p < 0.01 vs model group
Discussion
Elevated cholesterol is an important risk factor for atherosclerotic cardiovascular disease, and strategies to reduce cholesterol levels have been used to reduce the risk of atherosclerotic cardiovascular disease. Ezetimibe, a clinically used cholesterol absorption inhibitor, works via inhibiting the function of NPC1L1 [4]. In this LDLR−/− hamster study, we report for the first time that: 1) ezetimibe significantly promoted the protein expression of CYP7A1 and PPARγ, and lowered the protein expression of PPARα and PPARβ in the liver; 2) ezetimibe significantly up-regulated the protein expression of LXRβ in the small intestine, but not NPC1L1 and ABCG5/G8; and 3) ezetimibe may not influence PCSK9 in the absent of LDLR. Given that this study was performed in the animal model lacking LDLR, these data may be more relevant for humans with familial hypercholesterolemia than for general population.
As previously reported by Guo et al., high-fat diet can induce human-like hyperlipidemia in LDLR−/− hamsters [17]. The TC-lowering effect of ezetimibe was consistent with the results obtained from C57BL/6 J mice [13], apolipoprotein E knockout mice [24], rhesus monkeys [6], and humans [10]. The mechanisms may be attributed to the significant decreased dietary cholesterol absorption (~ 86% in C57BL/6 J mice) and the increased RCT from peripheral tissue macrophages [13]. The TG-lowering effect of ezetimibe in LDLR−/− hamsters was inconsistent with the data in rhesus monkeys [6], and humans [10]. However, our data were consistent with the previous results obtained in LDLR−/− animals [17, 19], which also indicated that ezetimibe not only significantly reduced plasma TC levels but also decreased plasma TG levels. These differences may be attributed to the distinct animal models, and especially the absent of LDLR. Given that ezetimibe dose not block fat absorption [25], the activity of ezetimibe in decreasing TG is likely a secondary effect of diminished cholesterol delivery in LDLR−/− mice or hamsters [19, 26]. Due to the significant difference in lipid profile between the ezetimibe and model group and especially the small variation within each group, 3 of the 5 hamsters per group were randomly chosen for the immunoblotting experiments in the present study.
The LDL-C lowering effect of ezetimibe is usually attributed primarily to increased catabolism of LDL-C via up-regulation of LDLR [19, 27]. The changes of LDL-C in LDLR−/− models can directly refect the rate of LDL-C production. Our data together with the previous publication [19] support that ezetimibe treatment can decrease LDL production. Given the absence of LDLR in LDLR−/− hamsters, SR-B1 plays a more important role in the hepatic uptake of cholesterol from plasma. However, our data showed that ezetimibe had no effect on the expression of SR-B1, which was consistent with a previous study in LDLR−/− mice [19]. Additionally, we report here that ezetimibe treatment has no modulatory effect on the protein expression of PCSK9, which functions as a negatively modulator of LDLR [23]. Cholesterol in the liver can be transformed to bile acid and then excreted into bile or secreted back into plasma within VLDL. CYP7A1 is a key rate-limiting enzyme for the transformation from cholesterol to bile acid. In a previous study, ezetimibe seemed to increase the mRNA expression of CYP7A1 based on the treatment of ursodiol in mice [15] and in germ-free or specific pathogen-free mice [28]. In this study, we report for the first time that ezetimibe significantly increased the protein expression of CYP7A1 in LDLR−/− hamsters. This result suggested that ezetimibe may accelerate RCT by enhancing the transformation from cholesterol to bile acid. A previous study has successfully demonstrated that ezetimibe enhanced RCT via the hepato-biliary pathway in hamsters [29].
Excretion of cholesterol by the liver and intestine is mediated by the tissue-specific expression of the transporters ABCG5 and ABCG8, which transfer these sterols into the lumen of the biliary tree or intestine, respectively [14, 30]. The ABCG5/G8 heterodimer account for ~ 70 to 90% of biliary cholesterol secretion [31]. In addition to its known suppression of intestinal cholesterol absorption, one previous study demonstrated that ezetimibe can stimulate macrophage-to-feces RCT by indirectly increasing liver ABCG5/G8 mRNA expression, but not in the small intestine [14]. However, another study indicated that ezetimibe increased fecal neutral sterol elimination via ABCG5/G8 independent manner as verified in ABCG5/G8-deficient mice [15]. Furthermore, transgenic mice expressing human NPC1L1 in hepatocytes resulted in 10- to 20-fold decrease in biliary cholesterol concentration, but it showed no effect on the protein expression of ABCG5/G8, SR-B1 and LDLR [32]. In a diet-induced hamster model of insulin resistance, ezetimibe also showed no effect on the protein expression of ABCG5 and ABCG8 [33]. Our data were consistent with the previous studies, which showed ezetimibe treatment has no significant effects on the hepatic and intestinal ABCG5/G8 protein expression. LXRα is the best-studied regulator of ABCG5/G8 expression, however, our results in LDLR−/− hamsters and the previous study in C57BL/6 J mice [14] all demonstrated that ezetimibe treatment may not activate LXRα. SREBP-2 is also a regulator of ABCG5 expression in the liver [34], and our data suggested that ezetimibe showed no effect on SREBP-2. Furthermore, the loss of LXRβ seems to have no effect on the expression of ABCG5/G8 [35]. This may explain why there was a significant upregulation of LXRβ, but the protein expression of ABCG5/G8 showed no significant difference in the small intestine of the LDLR−/− hamsters.
In humans, ezetimibe treatment enhanced de novo cholesterol synthesis to maintain cholesterol balance. SREBP-2 can activate a series of genes involved in the synthesis of cholesterol, while SREBP-1c plays a key role in TG synthesis. Our data showed that ezetimibe administration had no significant effect on these two important lipid synthesis modulators. Furthermore, PPARα is highly expressed in the liver and its activation can promote the utilization of fat, while PPARγ is a key regulator of adipogenesis [21, 22]. Our data suggested that ezetimibe may enhance TG storage by reducing PPARα and promoting PPARγ expression in response to the significant reduction of TG in ezetimibe-treated LDLR−/− hamsters. These kinds of compensatory effects are also found in other ezetimibe administration studies, for example, the up-regulation of hydroxymethylglutaryl-CoA reductase (HMG-CoA) reductase and HMG-CoA synthase [19, 27]. However, ezetimibe showed no effect on the mRNA expression of HMG-CoA reductase and HMG-CoA synthase in C57BL/6 mice [15]. Therefore, the compensatory effects of ezetimibe seem to be different based on distinct animal species.
Finally, it is worthy to note that ezetimibe showed no effect on intestinal NPC1L1 in this study, and the result was consistent with a previous study in high-fat diet hamster [32]. In humans, only about 25–35% of intestinal cholesterol is derived from diet, while the remaining originates from the liver in bile, and perhaps a direct transintestinal cholesterol efflux (TICE) route [11, 12]. Presently, there are still arguments about the role of TICE pathway in promoting macrophage RCT [3]. However, accumulating evidence support that the absorption of cholesterol from the intestine is also an important determinant of macrophage-to-feces RCT [13, 36]. A recent study indicated that TICE may contribute minor to the macrophage-to-feces RCT in hamsters [29]. Unlike humans, hamsters express comparably lower NPC1L1 in the liver [37] as that of mice, which is a limitation of this animal model.
Conclusions
Ezetimibe significantly lowers TC and TG in LDLR−/− hamsters. It may improve RCT by up-regulating CYP7A1, but not ABCG5/G8. As a compensatory mechanism, it down-regulates PPARα and β and up-regulates PPARγ. However, these results need to be confirmed by other animal models.
Availability of data and materials
The authors made reproducible materials described in the manuscript, freely available to any scientist wishing to use them, without breaching participant confidentiality. The date and methods have been fully presented in the manuscript.
Abbreviations
- ABC:
-
ATP-binding cassettes;
- CYP7A1:
-
Cholesterol 7 alpha-hydroxylase A1
- HDL:
-
High density lipoprotein
- LDL:
-
Low density lipoprotein
- NPC1L1:
-
Niemann-Pick C1-like 1
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- PPAR:
-
Proliferator-activated receptor
- RCT:
-
Reverse cholesterol transport
- SR-BI:
-
Scavenger receptor B type 1
- SREBP:
-
Sterol regulatory element-binding protein
References
Nanna MG, Navar AM, Wojdyla D, Peterson ED. The association between low-density lipoprotein cholesterol and incident atherosclerotic cardiovascular disease in older adults: results from the national institutes of health pooled cohorts. J Am Geriatr Soc. 2019. https://doi.org/10.1111/jgs.16123.
Wang DQ. Regulation of intestinal cholesterol absorption. Annu Rev Physiol. 2007;69:221–48.
Xie P, Jia L, Ma Y, Qu J, Miao H, Wang N, Guo F, Yazdanyar A, Jiang XC, Yu L. Ezetimibe inhibits hepatic Niemann-pick C1-like 1 to facilitate macrophage reverse cholesterol transport in mice. Arterioscler Thromb Vasc Biol. 2013;33(5):920–5.
Carcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR Jr, Dean DC, Detmers PA, Graziano MP, Hughes M, Macintyre DE, Ogawa A, O'neill KA, Iyer SP, Shevell DE, Smith MM, Tang YS, Makarewicz AM, Ujjainwalla F, Altmann SW, Chapman KT, Thornberry NA. The target of ezetimibe is Niemann-pick C1-like 1 (NPC1L1). Proc Natl Acad Sci U S A. 2005;102(23):8132–7.
Sudhop T, Lutjohann D, Kodal A, Igel M, Tribble DL, Shah S, Perevozskaya I, von Bergmann K. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circ. 2002;106(15):1943–8.
van Heek M, Compton DS, Davis HR. The cholesterol absorption inhibitor, ezetimibe, decreases diet-induced hypercholesterolemia in monkeys. Eur J Pharmacol. 2001;415(1):79–84.
Lin X, Racette SB, Ma L, Wallendorf M, Ostlund RE Jr. Ezetimibe increases endogenous cholesterol excretion in humans. Arterioscler Thromb Vasc Biol. 2017;37(5):990–6.
Jakulj L, van Dijk TH, de Boer JF, Kootte RS, Schonewille M, Paalvast Y, Boer T, Bloks VW, Boverhof R, Nieuwdorp M, Beuers UH, Stroes ES, Groen AK. Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab. 2016;24(6):783–94.
Ouimet M, Barrett TJ, Fisher EA. HDL and reverse cholesterol transport. Circ Res. 2019;124(10):1505–18.
Davidson MH, Voogt J, Luchoomun J, Decaris J, Killion S, Boban D, Glass A, Mohammad H, Lu Y, Villegas D, Neese R, Hellerstein M, Neff D, Musliner T, Tomassini JE, Turner S. Inhibition of intestinal cholesterol absorption with ezetimibe increases components of reverse cholesterol transport in humans. Atheroscler. 2013;230(2):322–9.
van der Velde AE, Brufau G, Groen AK. Transintestinal cholesterol efflux. Curr Opin Lipidol. 2010;21:167–71.
Yu XH, Zhang DW, Zheng XL, Tang CK. Cholesterol transprot system: an integrated cholesterol transport model involved in atherosclerosis. Prog Lipid Res. 2018;73:65–91.
Sehayek E, Hazen SL. Cholesterol absorption from the intestine is a major determinant of reverse cholesterol transport from peripheral tissure macrophages. Arterioscler Thromb Vasc Biol. 2008;28(7):1296–7.
Altemus JB, Patel SB, Sehayek E. Liver-specific induction of Abcg5 and Abcg8 stimulates reverse cholesterol transport in response to ezetimibe treatment. Metab. 2014;63(10):1334–41.
Wang Y, Liu X, Pijut SS, Li J, Horn J, Bradford EM, Leggas M, Barrett TA, Graf GA. The combination of ezetimibe and ursodiol promotes fecal sterol excretion and reveals a G5G8-independent pathway for cholesterol elimination. J Lipid Res. 2015;56(4):810–20.
Temel RE, Sawyer JK, Yu L, Lord C, Degirolamo C, McDaniel A, Marshall S, Wang N, Shah R, Rudel LL, Brown JM. Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab. 2010;12(1):96–102.
Guo X, Gao M, Wang Y, Lin X, Yang L, Cong N, An X, Wang F, Qu K, Yu L, Wang Y, Wang J, Zhu H, Xian X, Liu G. LDL receptor gene-ablated hamsters: a rodent model of familial hypercholesterolemia with dominant inheritance and diet-induced coronary atherosclerosis. EBioMed. 2018;27:214–24.
Wang J, He K, Yang C, Lin X, Zhang X, Wang Y, Liu G, Xian X. Dietary cholesterol is highly associated with severity of hyperlipidemia and atherosclerotic lesions in heterozygous LDLR-deficient hamsters. Int J Mol Sci. 2019;20(14) pii: E3515.
Repa JJ, Turley SD, Quan G, Dietschy JM. Delineation of molecular changes in intrahepatic cholesterol metabolism resulting from diminished cholesterol absorption. J Lipid Res. 2005;46(4):779–89.
Guo S, Tian H, Dong R, Yang N, Zhang Y, Yao S, Li Y, Zhou Y, Si Y, Qin S. Exogenous supplement of N-acetylneuraminic acid ameliorates atherosclerosis in apolipoprotein E-deficient mice. Atheroscler. 2016;251:183–91.
Yin J, Wang J, Li F, Yang Z, Yang X, Sun W, Xia B, Li T, Song W, Guo S. The fucoidan from the brown seaweed Ascophyllum nodosum ameliorates atherosclerosis in apolipoprotein E-deficient mice. Food Funct. 2019;10(8):5124–39.
Yang Z, Liu G, Wang Y, Yin J, Wang J, Xia B, Li T, Yang X, Hou P, Hu S, Song W, Guo S. Fucoidan A2 from the brown seaweed Ascophyllum nodosum lowers lipid by improving reverse cholesterol transport in C57BL/6J mice fed a high-fat diet. J Agric Food Chem. 2019;67(20):5782–91.
Wu MF, Xu KZ, Guo YG, Yu J, Wu Y, Lin LM. Lipoprotein(a) and atherosclerotic cardiovascular disease: current understanding and future perspectives. Cardiovasc Drugs Ther. 2019. https://doi.org/10.1007/s10557-019-06906-9.
Davis HR Jr, Compton DS, Hoos L, Tetzloff G. Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2001;21(12):2032–8.
van Heek M, Farley C, Compton DS, Hoos L, Davis HR. Ezetimibe selectively inhibits intestinal cholesterol absorption in rodents in the presence and absence of exocrine pancreatic function. Br J Pharmacol. 2001;134(2):409–17.
van Heek M, Austin TM, Farley C, Cook JA, Tetzloff GG, Davis HR. Ezetimibe, a potent cholesterol absorption inhibitor, normalizes combined dyslipidemia in obese hyperinsulinemic hamsters. Diab. 2001;50(6):1330–5.
Tremblay AJ, Lamarche B, Cohn JS, Hogue JC, Couture P. Effect of ezetimibe on the in vivo kinetics of apoB-48 and apoB-100 in men with primary hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2006;26(5):1101–6.
Zhong CY, Sun WW, Ma Y, Zhu H, Yang P, Wei P, Zeng BH, Zhang Q, Liu Y, Li WX, Chen Y, Yu L, Song ZY. Microbiota prevents cholesterol loss from the body by regulating host gene expression in mice. Sci Rep. 2015;5:10512.
Uto-Kondo H, Ayaori M, Sotherden GM, Nakaya K, Sasaki M, Yogo M, Komatsu T, Takiguchi S, Yakushiji E, Ogura M, Nishida T, Endo Y, Ikewaki K. Ezetimibe enhances macrophage reverse cholesterol transport in hamsters: contribution of hepato-biliary pathway. Biochim Biophys Acta. 2014;1841(9):1247–55.
Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH. Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest. 2002;110(5):671–80.
Sabeva NS, Liu J, Graf GA. The ABCG5 ABCG8 sterol transporter and phytosterols: implications for cardiometabolic disease. Curr Opin Endocrinol Diab Obes. 2009;16:172–7.
Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann-pick C1-like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. 2007;117(7):1968–78.
Naples M, Baker C, Lino M, Iqbal J, Hussain MM, Adeli K. Ezetimibe ameliorates intestinal chylomicron overproduction and improves glucose tolerance in a diet-induced hamster model of insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2012;302(9):G1043–52.
Temel RE, Brown JM. Biliary and nonbiliary contributions to reverse cholesterol transport. Curr Opin Lipidol. 2012;23:85–90.
Kim HJ, Fan X, Gabbi C, Yakimchuk K, Parini P, Warner M, Gustafsson JA. Liver X receptor beta (LXRbeta): a link between beta-sitosterol and amyotrophic lateral sclerosis-Parkinson’s dementia. Proc Natl Acad Sci U S A. 2008;105(6):2094–9.
Briand F, Naik S, Fuki I, Millar JS, Macphee C, Walker M, Billheimer J, Rothblat G, Rader DJ. Both the peroxisome proliferator-activated receptor delta agonist, GW0742, and ezetimibe promote reverse cholesterol transport in mice by reducing intestinal reabsorption of HDL-derived cholesterol. Clin Transl Sci. 2009;2(2):127–33.
Valasek MA, Repa JJ, Quan G, Dietschy JM, Turley SD. Inhibiting intestinal NPC1L1 activity prevents diet-induced increase in biliary cholesterol in Golden Syrian hamsters. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G813–22.
Acknowledgements
We thank professor Chenfeng Ji at Harbin University of Commerce for providing useful suggestions.
Funding
This work was supported by the National Natural Science Foundation of China (81770463 and 31300639); the Shandong Provincial Natural Science Fund (ZR2013HQ014); Shandong Science and Technology Development Funding of Traditional Chinese Medicine (2019414) and the Scientific Foundation of Weifang Medical University.
Author information
Authors and Affiliations
Contributions
All authors participated in the collection and analysis of the date. B Xia, P Lin, J Yin, and Z Yang were responsible for the western blot study and data statistical analysis. J Wang, X Yang and T Li were responsible for the animal daily care and sampling. Y Ji, F Li and S Guo were responsible for the design and funding of this study. S Guo was responsible for manuscript preparation. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal experiments were approved by the Laboratory Animal Ethical Committee of Weifang Medical University and followed the NIH guidelines for the care and use of animals.
Consent for publication
All authors agree to publish this article in the journal of Lipids in Health and Disease.
Competing interests
The authors declare that there are no conflicts of interest in the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xia, B., Lin, P., Ji, Y. et al. Ezetimibe promotes CYP7A1 and modulates PPARs as a compensatory mechanism in LDL receptor-deficient hamsters. Lipids Health Dis 19, 24 (2020). https://doi.org/10.1186/s12944-020-1202-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-1202-5