
Abstract
Using an established CRISPR-Cas mediated genome editing technique for streptomycetes, we explored the combinatorial biosynthesis potential of the auroramycin biosynthetic gene cluster in Streptomyces roseosporous. Auroramycin is a potent anti-MRSA polyene macrolactam. In addition, auroramycin has antifungal activities, which is unique among structurally similar polyene macrolactams, such as incednine and silvalactam. In this work, we employed different engineering strategies to target glycosylation and acylation biosynthetic machineries within its recently elucidated biosynthetic pathway. Auroramycin analogs with variations in C-, N- methylation, hydroxylation and extender units incorporation were produced and characterized. By comparing the bioactivity profiles of five of these analogs, we determined that unique disaccharide motif of auroramycin is essential for its antimicrobial bioactivity. We further demonstrated that C-methylation of the 3, 5-epi-lemonose unit, which is unique among structurally similar polyene macrolactams, is key to its antifungal activity.
Similar content being viewed by others
Introduction
Natural products (NPs) are an important source for pharmacological applications with a significant proportion of current drugs being natural product or natural product-derived [1]. Advances in genome editing and synthetic biology tools, together with natural product biosynthesis knowledge accumulated over the decades, allow us to better predict, design and build pathways towards the synthesis of natural products [2]. Earlier, we established a rapid and efficient CRISPR-Cas9 strategy for biosynthetic gene cluster (BGC) editing and activation in streptomycetes [3, 4], which opens up opportunities for combinatorial biosynthesis in native streptomycete hosts [5]. Compared to chemical syntheses, combinatorial engineering of native biosynthetic pathways allows us to design and biosynthesize structurally complex chemical analogs without traversing difficult multi-step and possibly low-yielding chemical reactions, thus facilitating elucidation of structure–activity relationships towards an optimized drug lead.
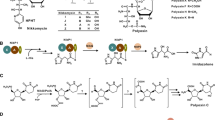
In this study, we describe our efforts to engineer the BGC of antimicrobial auroramycin (Fig. 1a, [6, 7]) and characterize its structure activity relationship (SAR). Auroramycin (1) is a polyene macrolactam that is doubly glycosylated. Sugars are attached in the order of xylosamine and 3, 5-epi-lemonose to the polyketide core. Compared to structurally similar natural products (Fig. 1b), such as the doubly glycosylated incednine [8] and monoglycosylated silvalactam [9], auroramycin is the only polyene macrolactam with reported antifungal activity to date. One of the main structural differences between auroramycin, incednine and silvalactam is their glycosylation pattern (Fig. 1b). Glycosylation can significantly increase the diversity and complexity of natural products and has often been shown to directly and significantly impact the their bioactivity and pharmacological properties [10, 11]. As such, glycodiversification is an attractive strategy to diversify and optimize bioactivity of NPs. Auroramycin also has a unique isobutyrylmalonyl (ibm) moiety incorporated into its polyene core. This ibm extender unit is relatively rare among NPs [12]. As a mixture of malonyl and methylmalonyl moieties are also incorporated into the polyketide, acyltransferase engineering would also allow us to quickly access new structurally diverse analogs.

a Biosynthetic gene cluster and b structures of auroramycin, its analogs produced in this study, along with structurally related polyene macrolactams, incednine and silvalactam. The different gene manipulations on the BGC are annotated by *. Blue * for promoter insertion, red * for site mutation and black * for in-frame gene deletion. Operons for the various biosynthetic pathways are also annotated and color-coded; red is for glycosylation and sugar biosynthesis pathways, green is for extender unit biosynthesis, blue is for polyketide synthases and purple is for starter unit biosynthesis. The gene table corresponding to the gene cluster is also available as Additional file 1: Table S1 and detailed biosynthetic schemes can be found in [6]
Here, we describe pathway engineering for glycosylation and extender unit incorporation in native S. roseosporous. To examine importance of polyketide backbone modifications and its unique disaccharide moiety towards anti-MRSA and antifungal activities, five new auroramycin analogs were produced and characterized (Fig. 1).
Results
Engineering sugar biosynthesis
We previously proposed the sugar biosynthesis pathways for auroramycin (Fig. 2a, [6]). Based on this, deletion of the N,N-dimethyltransferase AurS9 and C-methyltransferase AurS11 should remove N-methylation and C-methylation of 3, 5-epi-lemonose to yield compounds 3 and 4 respectively. As expected, 3 and 4 were main products of the respective engineered strains (Figs. 1b, 2b). Production yields of 3 and 4 were 40–80 mg/L, which are comparable to that of auroramycin. The latter observation suggested that the glycosyltransferase for 3, 5-epi-lemonose was able to transfer different non-methylated sugars onto the auroramycin scaffold just as efficiently as its cognate sugar substrate.

Engineering 3, 5-epi-lemonose biosynthesis. a Putative sugar biosynthesis pathway. b Liquid chromatography mass spectrometry (LCMS) spectra of extracts from engineered S. roseosporus strains with no modification to native BGC, (top) deletion of aurS9 and (middle) deletion of aurS11 (bottom) in the auroramycin BGC
Engineering glycosylation
Two glycosylation events take place during auroramycin biosynthesis. However within the BGC, there are four annotated glycosyltransferases (AurS4, S5, S10, S13) in addition to a P450 (AurS12) as its potential auxiliary partner, DesVIII [13, 14]. The four glycosyltransferases have high similarity to DesVII/EryCIII glycosyltransferases which require activation and stabilization by an DesVIII homolog auxiliary partner. A DesVII/DesVIII complex is required for activity. Of these four glycosyltransferases, two were truncated and hypothesized to be inactive; N-terminal helices, hypothesized for DesVIII interactions, and potential sites for substrates and sugar interactions were missing or modified (Additional file 1: Figure S9, [13, 14]). Earlier, we obtained aglycon 2 by deleting a 13 kb region in the BGC, which includes the aurS5, S10, S12, S13 genes [6]. To assign the roles of individual glycosyltransferases and putative auxiliary partner, as well as produce the mono-glycosylated analog 5 for SAR studies, we systematically deleted each of the genes and characterized the products of the engineered S. roseosporus strains (Fig. 3).

Engineering glycosylation. a LCMS spectra of engineered S. roseosporus strains with 13 kb and aurS4 deletions within the BGC. b LCMS spectra of engineered S. roseosporus strains with no deletion, aurS10, aurS12 and aurS13 deletions within the BGC. c Table depicting the genes that were deleted, their annotated gene products and resulting metabolite produced by S. roseosporus strains carrying the deletion. Length refers to the number of amino acid residues of the indicated gene product. d Putative glycosylation scheme of auroramycin. AurS10 is not essential for auroramycin glycosylation but increases the efficiency of the second glycosylation reaction
Individual deletions of the 5 genes involved in auroramycin glycosylation revealed that different sets and interactions of glycosyltransferases are required for each of the two glycosylation events (Fig. 3a–c). The first glycosylation event required the gene products of aurS4 and aurS5 as deletion of either gene yielded aglycon 2 (Fig. 3a). The second glycosylation event likely involved a more traditional DesVII/VIII complex encoded by aurS12 and aurS13. Purification and characterization of co-expressed AurS12 and AurS13 revealed that AurS13 and AurS12 have similar oligomerization profile as the DesVII/DesVIII complex [13, 14], suggesting that the two proteins form a functional glycosylation complex similar to the latter (Additional file 1: Figure S10). Deletion of either aurS12 and aurS13 yielded the mono-glycosylated analog 5 (Fig. 3b). As aglycon 2 was not observed with aurS12 deletion, this suggested that the first glycosylation step did not require an auxiliary protein partner for activity. AurS10 was also not essential for auroramycin glycosylation but most likely enhanced the second glycosylation event since deletion of aurS10 resulted in the production of a mixture of analog 5 and auroramycin (Fig. 3b).
Based on these observations, we proposed the following scheme for glycosylation in the auroramycin biosynthetic pathway: xylosamine is first glycosylated by AurS4 and AurS5, after which, 3, 5-epi-lemonose is added by AurS12 and AurS13 with AurS10 needed for increased efficiency of the second glycosylation step (Fig. 3c, d). Notably, contrary to in silico predictions that the truncated gene products of aurS4 and aurS10 are non-functional (Additional file 1: Figure S9, [15]), deletion of AurS4 and AurS10 has a profound effect on auroramycin’s glycosylation, suggesting a functional role for these truncated gene products.
Engineering extender unit
To engineer the extender units that constitute the macrocycle core (Figs. 1, 4), we first examined a strategy consisting of complementation of an inactivated cis-acyltransferase with a trans-acting acyltransferase of a different specificity (Fig. 4a, b, [16]). Previous AT complementation examples include complementation of a single module (DEBS Mod6) for the production of 2-desmethyl-6-dEB by malonyltransferase [17] and by a trans-acting AT from bryostatin PKS [18]. These studies demonstrated targeted incorporation of malonyl CoA (mCoA) into the polyketide carbon backbone. To engineer the macrocycle core of auroramycin, we chose a highly active malonyl CoA-specific trans-acting acyltransferase from the disorazole PKS (Dszs AT, [19]) to functionally rescue inactivated methylmalonyl CoA (mmCoA)-specific and ibmCoA-specific acyltransferases in the auroramycin PKS (Fig. 4b).

Extender unit engineering of the auroramycin PKS. a Polyketide synthase assembly. b AT complementation schematic (left: native AT in modular PKS, right, complementation of inactivated AT by trans-acting AT). c LCMS spectra of the auroramycin-producing strain compared to AT6°, AT9° and AT10° strains complemented by dszsAT. KS ketosynthase, AT acyltransferase, KR ketoreductase, ACP acyl carrier protein, DH dehydratase, ER enoyl reductase, TE thioesterase, mAT malonyl CoA-specific acyltransferase
Dszs AT complementation of inactive AT6 and AT9 (AT6°, AT9°) yielded the expected products 6 and 7 respectively (Figs. 4c, 1b). However, yields differed significantly with 6 produced at 50% yield compared to 7 (Fig. 4c). The latter observation was most likely due to substrate specificities of the downstream modules that limited their abilities to process the non-native intermediates [20,21,22]; the intermediate of AT6° required processing along four modules compared to the AT9° intermediate, which had to be accepted by a single module before cyclization.
With Dszs AT complementation of inactive AT10 (AT10°), instead of obtaining only the malonyl moiety incorporated compound (8, Fig. 4c), we also found the mmCoA analog 9. Analogs 8 and 9 were produced at an approximately 1:2 ratio with yields less than 5% of auroramycin. Substrate preference of Dszs AT for mCoA to mmCoA was observed previously to be approximately more than 46,000-fold [23]. Thus the significant decrease in product yields, along with the observed mmCoA-incorporated product, suggested that C-2 methylation is highly favoured by downstream gatekeeper domains, in particular the thioesterase domain [24]. Due to the low yields of analogs 8 and 9, their bioactivity was not characterized.
Post PKS hydroxylation
Among structurally similar natural products such as incednine, silvalactam and auroramycin (Fig. 1b), post-PKS hydroxylation takes place at a methylated site on the polyene core carbon skeleton. Conservation of this functional group suggests that either hydroxylation or/and methylation at this site might be important for bioactivity. To investigate the role of the additional hydroxyl group at C-10, we examined the production of dehydroxylated analog 10. In the aurO1 deletion mutant, only production of the dehydroxylated analog 10 was observed (Fig. 5).

Post PKS hydroxylation. a Proposed hydroxylation reaction and b respective LCMS of the engineered S. roseosporus in comparison to the auroramycin producing strain
Antimicrobial activities of auroramycin analogs
The antifungal activities of five analogs generated in this study were compared to auroramycin against six fungal and yeast species (Table 1). Analogs 3 and 4, which had perturbations in sugar biosynthesis, gave drastically different results in their bioactivities. The de-N-methylated analog 3 retains most of its bioactivity compared to auroramycin. Notably, new bioactivity (against Kodameae ohmeri) was observed with analog 3, suggesting that removing the N-methylation on the 3, 5-epi-lemonose in auroramycin may improve its potential as a broad spectrum antifungal. In contrast, analog 4 is inactive against the tested fungal and yeast strains, highlighting the importance of the C-methylation on the 3, 5-epi-lemonose in auroramycin. As predicted, the mono-glycosylated analog 5, which is most structurally similar to silvalactam lost all antifungal activity. Our results corroborated previous findings that silvalactam is inactive against yeast Saccharomyces cerevisiae [9], and further highlighted the importance of having the additional outer sugar for antifungal bioactivity. Interestingly, analogs 6 and 10, which contained modifications at C-10 of the macrolactam core, had differing outcomes in their bioactivities. Removal of the hydroxyl group at C-10 (analog 10) largely retained its bioactivity whereas removal of the methyl group at C-10 (analog 6) led to complete loss of bioactivity against the tested species.
We also tested antibacterial activities of the five auroramycin analogs against three Gram-positive bacteria strains, namely methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-intermediate methicillin-resistant Staphylococcus aureus (VI-MRSA) and vancomycin-resistant Enterococcus faecalis (VRE) (Table 2). While there were some notable trends for the antibacterial activities of the analogs, there were distinct differences between the antifungal and antibacterial activities exhibited by the auroramycin analogs. While the analogs generally exhibited either clear retention or complete loss of antifungal bioactivity, alterations to the antibacterial activities for the analogs were more modest. Analog 3 retained its antibacterial activity whereas analog 4 showed some loss of activity, especially against VI-MRSA. This reflected a similar but less drastic effect of the sugar biosynthesis perturbations on antibacterial activity compared to antifungal activity. Even without its outer 3, 5-epi-lemonose sugar, analog 5 retained partial antibacterial activity. While modifications at C-10 of the macrolactam core gave differing outcomes in analogs 6 and 10 antifungal bioactivities, antibacterial bioactivities for analogs 6 and 10 are generally significantly reduced. Like auroramycin, the analogs are inactive against Gram-negative bacteria (MIC > 128 μg/mL).
Discussion
Among structurally similar polyene macrolactams, auroramycin is the only one with reported antifungal activity to date. Furthermore, chemogenomic analyses in yeast also determined that auroramycin’s ergosterol-independent mode of action is distinct from common antifungal polyenes, such as amphotericin, nystatin, which are known to bind to ergosterols [7]. To identify the functional groups responsible for endowing auroramycin with antifungal and anti-MRSA activities, we sought to engineer auroramycin analogs and characterize the effect of specific chemical structural changes on their bioactivity profiles. Here we targeted sites unique to auroramycin (C, N-methylation on sugars) as well as sites that are common among auroramycin, silvalactam and incednine (unique extender units and hydroxylation). Through rational engineering and CRISPR-Cas mediated genome editing, we could rapidly design, build and test different S. roseopsorus mutants. In our study, production yields close to that of auroramycin were achieved for most of the analogs. This could be contributed to minimal disruption to the three-dimensional structure of the polyketide assembly line and sufficiently promiscuous cis-acting glycosylation and sugar biosynthesis enzymes. However, there are still engineering bottlenecks, as in the case of swapping the extender unit on C-2 on auroramycin, where minimal product was observed. To access analogs restricted by downstream gatekeeper domains, strategies such as directed evolution or rational design of downstream enzymes to increase substrate promiscuity will have to be examined [25, 26]. However, these strategies are not trivial and should be undertaken for natural products of high interest. Further diversity for targeted functional groups can be achieved by altering enzyme specificities and swapping enzyme domains [16, 27].
By comparing the bioactivity profiles of the five analogs that were achieved in good yields and purities, we were able to evaluate the functional importance of different chemical moieties on auroramycin (Fig. 6). From our studies, the unique disaccharide moiety was essential for antifungal and anti-MRSA bioactivity, however anti-MRSA activity can be restored with mono-glycosylation. Interestingly, our results demonstrated unexpected importance of the C-methylation on the outer sugar on bioactivity, especially for antifungal activity. The C-methylation on the outer sugar is unique to auroramycin and could explain auroramycin being the sole antifungal agent among similar polyene macrolactams. Perturbation of the macrolactam core of auroramycin led to mixed outcomes for antifungal activity but mostly resulted in loss of antibacterial activity. Overall, the structural features examined in our study defined auroramycin’s antifungal bioactivity more distinctly than its antibacterial activity. Additional modifications to the sugars and the degree of saturation of the polyene macrolactam core may be explored to improve the antifungal activity of auroramycin.

Structural activity map of auroramycin. Red shaded: removal resulted in drastic loss of both antifungal and antibacterial bioactivities; green shaded: removal resulted in retention of activity or slightly better antifungal activity, including new bioactivity against K. ohmeri; yellow shaded: minimal changes in antifungal activity but loss in potency in antibacterial activity; dashed blue circle: essential for antifungal activity; enhanced antibacterial potency; dotted red circle: due to the low production yields and purities, biological activity of these analogs were not functionally characterized
Due to highly efficient, precise and consistent CRISPR-Cas mediated genome editing and advanced DNA assembly methods [28, 29], we were able to accelerate the generation of 12 strains (with at least two genomic edits each) for analog production and screening in this study. Multiplex editing is expected to further accelerate strain construction [3]. Multiplex inactivation of key biosynthetic genes within a target BGC in streptomycetes can also be achieved with base editing using Cas9-deaminase fusions [30]. Last but not least, a major consideration of any genome editing strategy is that it requires the introduction of recombinant DNA, which may be challenging depending on the target actinomycete strain.
Conclusion
In this work, we used CRISPR-Cas mediated genome editing to accelerate the rational engineering of the antimicrobial auroramycin biosynthetic gene cluster. From a single design–build–test cycle, we were able to make specific changes to the glycosylation pattern and polyene macrolactam core of auroramycin to generate analogs. Five of these analogs were biosynthesized in good yields and their bioactivities were further characterized. By comparing the bioactivities of these analogs, we determined that the unique disaccharide moiety, in particular C-methylation of the outer sugar unit, was important for the antifungal activity of auroramycin.
Materials and methods
Growth and conjugation conditions
Unless otherwise indicated, all reagents are obtained from Sigma, St. Louis, USA. Conjugation experiments involving WM3780 E. coli strains were performed on R2 agar without sucrose. Unless otherwise indicated, strains are propagated in MGY medium at 30 °C. Spore preparations and conjugation protocols were similar as described before [3]. A typical spore prep contains ~ 106–107 spores/mL as determined by serial dilution plating. List of strains and plasmids used in this study is available in Additional file 1: Table S2.
Construction of genome editing plasmids
All DNA manipulations were carried out in OmniMAX™ (Thermo Fisher, Massachusetts, USA). Primers used in this study are listed in Additional file 2: Table S3. Restriction enzymes were obtained from New England Biolabs. Protospacers were first inserted via BbsI-mediated Golden Gate Assembly before introduction of the respective homology flanks via Gibson assembly, as previously described [3]. Detailed description of aurS5 deletion strain can also be found in [4].
Engineering of strains
Precise deletions of individual target genes without affecting intergenic regions were made using a CRISPR-Cas mediated editing strategy ([3], Additional file 1: Figure S1–S7). To delete aurS5, we had to use a different Cas protein [4]. As described elsewhere [6], in order to activate the entire BGC for production of the corresponding analogs, luxR in these edited strains was also placed under a strong constitutive kasO* promoter [31].
AT domains in modules 6, 9 and 10 were independently inactivated with a single active site serine to alanine mutation (Additional file 1: Figure S8). A gene cassette of Dszs AT, placed under kasO*p, was integrated into the attB site of the genome [32]. The luxR gene within the BGC in these strains was also under kasO*p to activate its production.
Validation of promoter knock-in and genome editing
Genomic DNA from wild type and exconjugants from the indicated strains were isolated from liquid cultures using the Blood and Tissue DNeasy kit (Qiagen, Hilden, Germany) after pretreating the cells with 20 mg/mL lysozyme for 0.5–1 h at 30 °C. PCR was performed using control primers beyond the homology regions with KODXtreme Taq polymerase (Millipore, Massachusetts, USA). Where indicated, PCR products were subjected to digest with specific restriction enzymes to differentiate between PCR products of wild type genomic sequences and successful genome editing by knock-ins. Positive samples were purified using ExoSAP-IT™ (Affymetrix USB, Massachusetts, USA) and validated by Sanger sequencing.
Fermentation
Fermentation of analogs and auroramycin was performed as described elsewhere [6] using the indicated engineered Streptomyces roseosporus NRRL 15998 strains.
Isolation and characterization of analogs
General considerations
Optical rotation was obtained on a JASCO P1030 polarimeter using a micro-cell (light path 1 cm). IR spectrum was taken on a PerkinElmer Spectrum 100 FT-IR spectrometer. UV spectra were recorded on Shimadzu UV-2450 UV–Vis spectrophotometer. CD spectra were taken on a JASCO J-810 CD Spectropolarimeter. HRMS spectra were measured on an Agilent 6545 Quadrupole-Time-of-Flight (Q-TOF) mass spectrometer. 1H, 13C, and 2D-NMR spectra were recorded on a Bruker 400 MHz and Varian VNMRS 700 MHz spectrometers and calibrated using residual non-deuterated solvent (CD2Cl2: δH = 5.32 ppm, δC = 53.84 ppm) as an internal reference.
Analog 3
To the crude ethyl acetate extracts from 200 agar fermentation plates (~ 6 L), cold ethyl acetate (20 mL × 2) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution and yellow solid. Cold methanol (10 mL) was then added to the yellow solid, sonicated and centrifuged to separate the yellow solution. The obtained white/pale yellow solid (238.0 mg, > 90% purity) was found to be mainly analog 3 together with monoglycosylated analog 5 in an approximate 7:1 ratio. See Additional file 1: Figure S11 for structural assignment for analog 3; HRMS m/z: 794.5000 [(M + H)+ calcd. for C44H68N3O8, 766.5006]. See latter for assignment of analog 5.
Analog 4
To the crude ethyl acetate extracts from 125 agar fermentation plates (~ 4 L), cold ethyl acetate (20 mL × 2) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution and yellow solid. Cold methanol (15 mL) was then added to the yellow solid, sonicated and centrifuged to separate the yellow solution and analog 4 was obtained as a white/pale yellow solid (315.0 mg, > 90% purity). [α]25D: − 301.6° (c 0.25, 1:1 CH2Cl2:MeOH); 1H and 13C NMR: see Additional file 1: Table S4; IR (KBr): ν = 3353, 2953, 2925, 2871, 1630, 1577, 1525, 1459, 1384, 1284, 1234, 1167, 1129, 1079, 1058, 985, 960 cm−1; UV/VIS (1:1 CH2Cl2:MeOH): λmax (log ε) = 317.5 (1.25) nm; HRMS m/z: 780.5161 [(M + H)+ calcd. for C45H70N3O8, 780.5163].
Analog 5
To the crude ethyl acetate extracts from 184 agar fermentation plates (~ 6 L), cold ethyl acetate (30 mL) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution and yellow solid. Cold methanol (5 mL) was then added to the yellow solid, sonicated and centrifuged to separate the yellow solution and analog 5 was obtained as a white/pale yellow solid (92.1 mg, > 94% purity). [α]25D: − 233.5° (c 0.2, 1:1 CH2Cl2:MeOH); 1H and 13C NMR: see Additional file 1: Table S4; IR (KBr): ν = 3350, 2925, 2854, 1712, 1628, 1530, 1463, 1386, 1285, 1147, 1077, 1057, 985, 961 cm−1; UV/VIS (MeOH): λmax (log ε) = 317.5 (1.74) nm; HRMS m/z: 623.4056 [(M + H)+ calcd. for C37H55N2O6, 623.4060].
Analog 6
To the crude ethyl acetate extracts from 200 agar fermentation plates (~ 4 L), cold diethyl ether (10 mL × 2) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution. Analog 6 was obtained in an approximate 17:1 ratio together with a hydroxylated product as a brown solid (107 mg, > 92% purity). See Additional file 1: Figure S12 for structural assignment; HRMS m/z: 780.5156 [(M + H)+ calcd. for C45H69N3O8, 780.5157].
Analog 7
To the crude ethyl acetate extracts from 191 agar fermentation plates (~ 4 L), cold diethyl ether (10 mL × 2) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution. Analog 7 was obtained in an approximate 5:1 ratio together with a hydroxylated product as a brown solid (165 mg, > 90% purity). See Additional file 1: Figure S13 for structural assignment; HRMS m/z: 780.5161 [(M + H)+ calcd. for C45H69N3O8, 780.5157]. Due to constraints of the sensitivity of the compounds to light and acid, further purification was not performed.
Analog 10
To the crude ethyl acetate extracts from 180 agar fermentation plates (~ 6 L), cold Et2O (10–15 mL × 2) was added, sonicated for 1–2 min and centrifuged to separate the yellow solution and brown solid. The brown solid was washed with cold 5:1 Et2O:acetone (2 mL × 3) and analog 10 was obtained as a brown solid (69.7 mg, > 94% purity). [α]25D: –261.9° (c 0.25, 1:1 CH2Cl2:MeOH); 1H and 13C NMR: see Additional file 1: Table S5; IR (KBr): ν = 3333, 2926, 2867, 1661, 1515, 1456, 1382, 1274, 1238, 1156, 1097, 1058, 988, 962 cm−1; UV/VIS (MeOH): λmax (log ε) = 317.5 (1.90) nm; HRMS m/z: 778.5361 [(M + H)+ calcd. for C46H72N3O7, 778.5370].
Antifungal assays
Measurements against indicated fungal strains were conducted at Eurofins Panlabs Taiwan (www.eurofins.com/epds), according to methodology described by the Clinical and Laboratory Standards Institute (CLSI) (M38-A, M27-A2).
Bacterial assays
Minimum inhibition concentration (MIC) values were determined by the broth microdilution method, as recommended by the Clinical and Laboratory Standards Institute with slight modifications. Briefly, purified auroramycin analogs were dissolved in DMSO, then diluted in Mueller–Hinton broth containing 0.2% DMSO. The organisms were tested at 5 × 105 CFU/mL. The MICs were read at 20 h after 35 °C incubation (Additional file 1: Figure S14).
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its Additional file 1].
Abbreviations
- SAR:
-
structure activity relationship
- Ibm:
-
isobutyrylmalonyl
- Dszs:
-
disorazole synthase
- PKS:
-
polyketide synthase
- KS:
-
ketosynthase
- AT:
-
acyltransferase
- KR:
-
ketoreductase
- ACP:
-
acyl carrier protein
- DH:
-
dehydratase
- ER:
-
enoyl reductase
- TE:
-
thioesterase
- mAT:
-
malonyl CoA-specific acyltransferase
- MRSA:
-
methicillin-resistant Staphylococcus aureus
- HRMS:
-
high resolution mass spectrometry
- LCMS:
-
liquid chromatography mass spectrometry
- mCoA:
-
malonyl Coenzyme A
- mmCoA:
-
methylmalonyl Coenzyme A
References
Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79(3):629–61.
Zhang MM, Qiao Y, Ang EL, Zhao H. Using natural products for drug discovery: the impact of the genomics era. Expert Opin Drug Discov. 2017;12(5):475–87.
Zhang MM, Wong FT, Wang Y, Luo S, Lim YH, Heng E, Yeo WL, Cobb RE, Enghiad B, Ang EL, Zhao H. CRISPR–Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nat Chem Biol. 2017;13(6):607.
Yeo WL, Heng E, Tan LL, Lim YW, Lim YH, Hoon S, Zhao H, Zhang MM, Wong FT. Characterization of Cas proteins for CRISPR-Cas editing in streptomycetes. Biotechnol Bioeng. 2019. https://doi.org/10.1002/bit.27021.
Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3(12):925.
Lim YH, Wong FT, Yeo WL, Ching KC, Lim YW, Heng E, Chen S, Tsai DJ, Lauderdale TL, Shia KS, Ho YS, Hoon S, Ang EL, Zhang MM, Zhao H. Auroramycin: a potent antibiotic from streptomyces roseosporus by CRISPR-Cas9 activation. ChemBioChem. 2018;19(16):1716–9.
Wong JH, Alfatah M, Kong KW, Hoon S, Yeo WL, Ching KC, Goh CJ, Zhang MM, Lim YH, Wong FT, Arumugam P. Chemogenomic profiling in yeast reveals antifungal mode-of-action of polyene macrolactam auroramycin. PLoS ONE. 2019;14(6):e0218189.
Futamura Y, Sawa R, Umezawa Y, Igarashi M, Nakamura H, Hasegawa K, Yamasaki M, Tashiro E, Takahashi Y, Akamatsu Y, Imoto M. Discovery of incednine as a potent modulator of the anti-apoptotic function of Bcl-xL from microbial origin. J Am Chem Soc. 2008;130(6):1822–3.
Schulz D, Nachtigall J, Geisen U, Kalthoff H, Imhoff JF, Fiedler HP, Süssmuth RD. Silvalactam, a 24-membered macrolactam antibiotic produced by Streptomyces sp. Tü 6392. J Antibiot. 2012;65(7):369.
Huang G, Lv M, Hu J, Huang K, Xu H. Glycosylation and activities of natural products. Mini Rev Med Chem. 2016;16(12):1013–6.
Elshahawi SI, Shaaban KA, Kharel MK, Thorson JS. A comprehensive review of glycosylated bacterial natural products. Chem Soc Rev. 2015;44(21):7591–697.
Lechner A, Wilson MC, Ban YH, Hwang JY, Yoon YJ, Moore BS. Designed biosynthesis of 36-methyl-FK506 by polyketide precursor pathway engineering. ACS Synth Biol. 2012;2(7):379–83.
Moncrieffe MC, Fernandez MJ, Spiteller D, Matsumura H, Gay NJ, Luisi BF, Leadlay PF. Structure of the glycosyltransferase EryCIII in complex with its activating P450 homologue EryCII. J Mol Biol. 2012;415(1):92–101.
Borisova SA, Liu HW. Characterization of glycosyltransferase DesVII and its auxiliary partner protein DesVIII in the methymycin/pikromycin biosynthetic pathway. Biochemistry. 2010;49(37):8071–84.
Takaishi M, Kudo F, Eguchi T. Identification of the incednine biosynthetic gene cluster: characterization of novel β-glutamate-β-decarboxylase IdnL3. J Antibiot. 2013;66(12):691.
Musiol-Kroll E, Wohlleben W. Acyltransferases as tools for polyketide synthase engineering. Antibiotics. 2018;7(3):62.
Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA: aCP transacylase. J Am Chem Soc. 2003;125(47):14307–12.
Lopanik NB, Shields JA, Buchholz TJ, Rath CM, Hothersall J, Haygood MG, Håkansson K, Thomas CM, Sherman DH. In vivo and in vitro trans-acylation by BryP, the putative bryostatin pathway acyltransferase derived from an uncultured marine symbiont. Chem Biol. 2008;15(11):1175–86.
Wong FT, Jin X, Mathews II, Cane DE, Khosla C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry. 2011;50(30):6539–48.
Jenner M, Frank S, Kampa A, Kohlhaas C, Pöplau P, Briggs GS, Piel J, Oldham NJ. Substrate specificity in ketosynthase domains from trans-AT polyketide synthases. Angew Chem Int Ed. 2013;52(4):1143–7.
Murphy AC, Hong H, Vance S, Broadhurst RW, Leadlay PF. Broadening substrate specificity of a chain-extending ketosynthase through a single active-site mutation. Chem Commun. 2016;52(54):8373–6.
Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, Okada M, Awakawa T, Ito T, Asakawa Y, Ueki M. Characterization of giant modular PKSs provides insight into genetic mechanism for structural diversification of aminopolyol polyketides. Angew Chem Int Ed. 2017;56(7):1740–5.
Dunn BJ, Watts KR, Robbins T, Cane DE, Khosla C. Comparative analysis of the substrate specificity of trans-versus cis-acyltransferases of assembly line polyketide synthases. Biochemistry. 2014;53(23):3796–806.
McMahon MD, Prather KL. Functional screening and in vitro analysis reveal thioesterases with enhanced substrate specificity profiles that improve short-chain fatty acid production in Escherichia coli. Appl Environ Microbiol. 2014;80(3):1042–50.
Zabala AO, Cacho RA, Tang Y. Protein engineering towards natural product synthesis and diversification. J Ind Microbiol Biotechnol. 2012;39(2):227–41.
Twigg F, Skyrud D, Li J, Zhang W. Engineering enzymes for natural product biosynthesis and diversification. Modern biocatalysis. Cambridge: Royal Society of Chemistry; 2018. p. 261–86.
Bernhardt P, O’Connor SE. Opportunities for enzyme engineering in natural product biosynthesis. Curr Opin Chem Biol. 2009;13(1):35–42.
Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4(5):e5553.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343.
Zhong Z, Guo J, Deng L, Chen L, Wang J, Li S, Xu W, Deng Z, Sun Y. Base editing in Streptomyces with Cas9-deaminase fusions. bioRxiv. 2019:630137.
Wang W, Li X, Wang J, Xiang S, Feng X, Yang K. An engineered strong promoter for streptomycetes. Appl Environ Microbiol. 2013;79(14):4484–92.
Combes P, Till R, Bee S, Smith MC. The Streptomyces genome contains multiple pseudo-attB sites for the φC31-encoded site-specific recombination system. J Bacteriol. 2002;184(20):5746–52.
Acknowledgements
The authors gratefully acknowledge the late Sydney Brenner for his discussion and support.
Funding
This work is supported by the Agency for Science, Technology and Research (A*STAR), Singapore and National Research Foundation, Singapore [NRF2013-THE001-094 to MMZ, FTW, YHL] and the A*STAR Visiting Investigator Program [1535j00137 to HZ].
Author information
Authors and Affiliations
Contributions
WLY performed and analysed conjugation and fermentation experiments. EH and LLT constructed plasmids for genetic manipulations. YWL, KCC, YHL performed chemical purifications and characterizations. D-JT, YWJ, T-LL, K-SS performed and analysed bacterial assays. HZ contributed to idea conception. ELA supervised WLY. MMZ contributed to idea conception and project design. YHL and FTW conceived project design, analysed data, prepared and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1. Table S1.
Putative functions of genes in the auroramycin biosynthetic gene cluster. Table S2. List of strains and plasmids used in this study. Table S4. 1H and 13C NMR data of auroramycin analog 4 and analog 5. Tabe S5. 1H and 13C NMR data of auroramycin analog 10. Figure S1. Alignment of sanger sequencing of aurS9 deletion mutant (edited) against wild type genome. Figure S2. Alignment of sanger sequencing of aurS11 deletion mutant (edited) against wild type genome. Figure S3. Alignment of sanger sequencing of aurS4 deletion mutant (edited) against wild type genome. Figure S4. Alignment of sanger sequencing of aurS10 deletion mutant (edited) against wild type genome. Figure S5. Alignment of sanger sequencing of aurS12 deletion mutant (edited) against wild type genome. Figure S6. Alignment of sanger sequencing of aurS13 deletion mutant (edited) against wild type genome. Figure S7. Alignment of sanger sequencing of aur01 deletion mutant (edited) against wild type genome. Figure S8. Alignment of sanger sequencing of AT mutant (edited) against wild type genome. Figure S9 Alignment of glycosyltransferases. Figure S10 Purification and characterization of His-tagged AurS13 expressed alone or His-tagged AurS13 co-expressed with AurS12 (no His-tag). Figure S11. Analytical data for the structural assignment of auroramycin analog 3. Figure S12. Analytical data for the structural assignment of auroramycin analog 6 and minor hydroxylated product. Figure S13. Analytical data for the structural assignment of auroramycin analog 7 and minor hydroxylated product. Figure S14. Representative microdilution assay plates for compound 1, 3-7, 10 with (A) Staphylococcus aureus N216, (B) S. aureus Z172, (C) Enterococcus faecalis ATCC 51299, (D) Pseudomonas aeruginosa ATCC 700603 and (E) E. coli ATCC 25922.
Additional file 2: Table S3.
Oligonucleotides used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yeo, W.L., Heng, E., Tan, L.L. et al. Biosynthetic engineering of the antifungal, anti-MRSA auroramycin. Microb Cell Fact 19, 3 (2020). https://doi.org/10.1186/s12934-019-1274-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-019-1274-y