
Abstract
Background
Mutations in voltage-gated sodium channel (SCN) genes are supposed to be of importance in the etiology of psychiatric and neurological diseases, in particular in the etiology of seizures. Previous studies report a potential susceptibility region at the chromosomal locus 2q including SCN1A, SCN2A and SCN3A genes for autism spectrum disorder (ASD). To date, there is no previous description of a patient with comorbid ASD and Tourette syndrome showing a deletion containing SCN2A and SCN3A.
Case presentation
We present the unique complex case of a 28-year-old male patient suffering from developmental retardation and exhibiting a range of behavioral traits since birth. He received the diagnoses of ASD (in early childhood) and of Tourette syndrome (in adulthood) according to ICD-10 and DSM-5 criteria. Investigations of underlying genetic factors yielded a heterozygous microdeletion of approximately 719 kb at 2q24.3 leading to a deletion encompassing the five genes SCN2A (exon 1 to intron 14–15), SCN3A, GRB14 (exon 1 to intron 2–3), COBLL1 and SCL38A11.
Conclusions
We discuss the association of SCN2A, SCN3A, GRB14, COBLL1 and SCL38A11 deletions with ASD and Tourette syndrome and possible implications for treatment.
Similar content being viewed by others


Background
Mutations in neuronal voltage gated sodium channel genes such as SCN1A, SCN2A and SCN3A are supposed to play an important role in psychiatric disorders and neurological diseases [1]. These three sodium channel genes encoding distinct α-subunit isoforms are clustered within 600 kB on chromosome 2q24 and are highly expressed in neurons and glia throughout the central and peripheral nervous system [2, 3].
Genome-wide scan studies for autism susceptibility genes reported a potential susceptibility region at locus 2q including SCN1A, SCN2A and SCN3A genes [4, 5].
Dravet-Syndrome (DS) is a childhood disorder associated with mainly loss-of-function mutations in the SCN1A gene. It is characterized by frequent seizures and devastating effects on cognitive and behavioural development, persisting into adulthood [6]. While approximately 70–80% of DS patients show mutations in SCN1A, some patients have variants in other genes including SCN2A [7].
Mutations of the SCN2A gene have also been reported in patients with autism [8] as well as with benign familial infantile seizures, intractable epilepsy, infantile spasms and severe myoclonic epilepsy of infancy [9,10,11,12].
Previous investigations mainly led to case reports with deletions on chromosome 2q24 being associated with seizures complicated by neurobehavioral comorbidities such as cognitive impairment, psychiatric disorders and social problems [13,14,15]. To date, there are only two published cases with deletions on chromosome 2q24 displaying a phenotype with autistic features and developmental delay, but no seizures: Celle et al. [2] described the case of a 3-year old boy presenting with autistic features, language delay, microcephaly and no history of seizures. He had an interstitial deletion of ~ 291.9 kb at band 2q24.3 with loss of the entire SCN2A gene and part of the SCN3A gene. Another case report described a 3 years and 4 months old girl with autistic features, developmental delay, mental retardation, language impairment and dysmorphic features, who carried a 2.8 Mb de novo deletion on chromosome 2q24.2 containing nine genes including SCN2A2, SCN3A, GRB14 and COBLL1 as well as LOC643397, FLJ39822, LOC643405, FIGN and TAIP-2 [16]. Until now, there is no description of a patient with SCN gene mutations and autism spectrum disorder (ASD) with additional symptoms of Tourette syndrome.
Case presentation
We present the unique case of a 28-year-old male patient displaying a complex clinical picture with mental retardation and various behavioural problems since birth. Symptoms of the autism spectrum comprising difficulties in social interaction and communication are reported since childhood. Additionally, he suffers from auto-aggressive tics in terms of beating himself with objects against his head and lower jaw, head movement tics and simple vocal tics. Striking dysmorphic features are not evident.
Developmental and somatic history
Except a one-time bleeding in week 20 of gestation, pregnancy had been without any complications. No infections, medication, smoking, or intake of alcohol or drugs during pregnancy was reported. The patient was delivered in week 40 of gestation with the help of a ventouse due to irregular cardiac activity. During delivery there were minor signs of birth asphyxia. Birth weight was 2.900 g (25th percentile), birth length 51 cm (50th percentile) and head circumference 33 cm (<3rd percentile). During infancy a prominent frontal fissure was conspicuous. A premature ossification of the sagittal fissure could not be detected.
The patient showed psychomotor retardation: he walked alone only by 26 months and was not able to sit alone falling over to one side without shoring up even at the age of 2. Furthermore, tics in terms of eye blinking as well as a muscular hypotonia were described. The patient’s parents reported early autistic features such as difficulties in social communication and interaction with avoiding eye contact and poor interest in social interaction. Development of speech was delayed (first words with 18 months). He refused body contact and demonstrated stereotypic patterns of behaviour such as filling bowls without showing any variations.
When examined at the age of 27 months, the patient presented some special facial features such as synophrys, epicanthus, modelled ears, a deep joined thumb and microcephaly. His weight was 10 kg (3rd percentile), his length 88 cm (25th percentile) and his head circumference 46 cm (2 cm above the 3rd percentile). The patient only spoke a few words and never a whole sentence. Mostly, he only repeated the words he had heard before in terms of an echolalia. A considerable general delay of development with severe perceptual disturbance and autistic traits was diagnosed.
Since the age of two years, the patient shows relevant aggressive symptoms such as throwing his head on the floor or biting into items. He needs extensive support concerning all activities of daily living and requires constant daily routines. During nursery school, auto-aggressive symptoms exacerbated in terms of head banging behaviour injuring his jaw and ears. At the age of 3 years, he started grinding his teeth. Later on, he presented head throwing movements against his left shoulder and banging of one row of his teeth against the other. There was a pattern of aggravation of the tic symptoms during stressful situations. He repeatedly showed refusal of meals and sleeping disturbances. At the age of 3 years, once there was a query febrile convulsion associated with an infection. Apart from that, there was no evidence for further seizures and clearly no epilepsy.
According to ICD-10 and DSM-5 classification, the clinical features of the patient are consistent with early infantile autism as well as with Tourette syndrome.
In the clinical examination, the patient presented with cauliflower-ears as a result of his head banging behaviour and subsequent repetitive ear injuries as well as various injuries of all kinds and healing stages. Dysmorphic features or additional external malformations were not noted, there was no evidence of internal abnormalities (heart, eye, inner ear) either. The patient showed a preserved ability of speech comprehension with rare speech production.
Family history
Our patient is the first son of non-consanguineous healthy German parents. His younger brother (24 years old) was diagnosed with an autism spectrum disorder (Asperger syndrome). There was no evidence of syndromal-secondary autism in the brother. He did not show intellectual impairment, dysmorphic signs or organic conditions such as a heart malformation or epilepsy. The mother’s paternal grandparents were cousins. They had five healthy children. In their further progeny three mentally retarded persons were reported, one granddaughter (died at the age of approximately 27 years) and two great-grandchildren (a boy and a girl). The exact diagnosis was unknown to our patient’s parents. Consanguinity of the retarded relatives’ parents was denied. One of the patient’s cousins (the son of his father’s brother) was said to suffer from clinically apparent dyslexia and perhaps an autism spectrum disorder. A disablement of a cousin of our patient’s father (son of his father’s sister) could not be specified further. An assessment of the father revealed no clinical characteristics, neither with respect to striking dysmorphic features, the occurrence of tics nor with respect to autistic symptoms. Further relatives with neurodevelopmental disorders were not reported on the father’s side of the family.
Cytogenetic and array-analysis
Conventional R-banded karyotyping of the patient was performed according to standard protocols with a resolution of approximately 500 bphs and revealed a structurally and numerically normal male karyotype (46,XY) in all 21 metaphases examined.
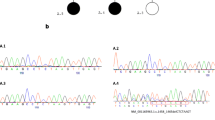
Furthermore, the genomic DNA of the patient was examined by microarray-analysis (CytoSureTM Constitutional v3 Array 180 k, OGT (Oxford Gene Technology)) according to the manufacturer’s instructions. After hybridization, the array was scanned with the SureScan Microarray scanner (Agilent), the results were analyzed using CytoSure interpret software v.4.9 (OGT) against the Genome Reference Consortium human genome GRCh37 (hg19). Molecular karyotyping revealed a heterozygous deletion of approximately 719 kb (267 contiguous oligonucleotides) out of the chromosomal region 2q24.3 (karyotype according to ISCN (International System for Human Cytogenetic Nomenclature) (2016): arr[GRCh37] 2q24.3(165471418_166190427)× 1). The deletion encompasses the five genes GRB14 (exon 1 to intron 2–3), COBLL1, SLC38A11, SCN3A and SCN2A (exon 1 to intron 14–15) listed in OMIM (Online Mendelian Inheritance in Man) (Fig. 1). With FISH (fluorescence in situ hybridization) analysis using probe RP11-150F4 (Empire Genomics) located at 2q24.3 the deletion could be confirmed. There is no evidence in literature that the microdeletion we detected is a common variant in the Caucasian population [17, 18]. “Orphanet” states a prevalence rate of a 2q24 microdeletion < 1/1000000 (worldwide) (https://www.orpha.net/data/patho/GB/uk-2q24.pdf).

Results of microarray-analysis. Molecular karyotyping revealed a deletion of approximately 719 kb out of the chromosomal region 2q24.3 encompassing the 5 genes GRB14 (exon 1 to intron 2–3), COBLL1, SLC38A11, SCN3A and SCN2A (exon 1 to intron 14–15) listed in OMIM (Online Mendelian Inheritance in Man)
The subsequently performed chromosome analysis of the parents, FISH with RP11-150F4 included, showed a normal female karyotype in the mother and a normal male karyotype in the father with no evidence of a deletion or rearrangement at 2q24.3. Therefore, it can be asserted that the deletion in the patient is a de novo mutation. The patient’s younger brother (suffering from Asperger syndrome) was shown to have a normal male karyotype without a deletion or rearrangement at 2q24.3.
Other investigations
Magnetic resonance imaging conducted in 2014 showed no abnormalities of the patient’s brain. In 1991, a detailed investigation was performed with a metabolic screening of serum, urine and cerebrospinal fluid revealing normal results. In the EEG, there was a slow baseline activity without any epilepsy suspicious potentials. A proton-spectroscopy showed no abnormalities of N-acetyl-aspartate, choline and phosphor-creatinine levels. An ophthalmologic investigation exhibited no deviations. In 1997, gastrointestinal passage was unsuspicious, and in an X-ray of the brain no stenosis of the sagittal fissure was confirmed.
Discussion and conclusions
In the present case report, we describe a 28-year-old male patient with symptoms of an early infantile autism as well as a Tourette syndrome. He carries a 719 kb deletion on chromosome 2q24.3 including the genes SCN3A and SCN2A which code for voltage-gated sodium channels. To date, no similar case has been described in the literature with respect to the phenotype of autism spectrum disorder plus Tourette syndrome but without epilepsy. The patient showed a global retardation of development (including psychomotor and language delay), various autistic traits, echolalia, severe auto-aggressive symptoms in terms of head banging, tic-specific head movements and vocal tics. There might have been one febrile convulsion at age 3; however, clearly there was no diagnosis of epilepsy. No other seizures had ever been reported. The somatic abnormalities included dysmorphic features such as synophrys, epicanthus, modelled ears, a deep joined thumb and microcephaly.
To date, there are only two case reports referring to patients with autistic features, language impairment, dysmorphic features and no history of seizures presenting a deletion of SCN2A and SCN3A genes [2, 16]. Chen et al. [16] described the most similar case to the one presented in this report. He presented a 3 years and 4 months old girl with autistic features, developmental delay, mental retardation, language impairment, dysmorphic features and no history of seizures with a 2.8 Mb de novo deletion on chromosome 2q24.2 entailing loss of nine genes including SCN2A2, SCN3A, GRB14 and COBLL1.
Bartnik et al. [13] identified a de novo ~ 110 kb deletion involving exons 1–2 of SCN2A and non-coding exon 1a of SCN3A in a 25-year-old female with mental retardation, neurobehavioural and psychiatric abnormalities and a history of infantile seizures. Additionally, there are several other case reports presenting patients with a deletion on chromosome 2q24 with current seizures or a history of seizures [13,14,15]. In all previous case reports, no additional symptoms of Tourette syndrome have been described to date.
The presently available literature suggests that mutations in SCN2A can be causative for ASD, benign familial neonatal infantile seizures, intractable epilepsy, infantile spasms and severe myoclonic epilepsy of infancy [8,9,10,11,12].
Since sodium channels are critical for action potential generation and propagation, a causative association between seizures and sodium channel dysfunction is plausible, whereas it is more difficult to understand this link for ASD and tics [1]. It is proposed that loss of function SCN1A mutations associated with seizures cause reduced GABA release leading to an inappropriate inhibition in related neuronal networks [6]. Parvalbumin neurons represent inhibitory GABAergic cells that are involved in various forms of feed-forward inhibition within the striatum [19]. Postmortem studies in patients with Tourette syndrome demonstrate a consistent and profound imbalance of parvalbumin-positive neuronal distribution in the basal ganglia [20]. The selective deficit of parvalbumin-positive and cholinergic striatal interneurons in Tourette syndrome was supposed to result in an impaired cortico/thalamic control of striatal neuron firing [21]. Evidence from studies demonstrates that Na(v)1.2 (the protein encoded by SCN2A) is abundant in parvalbumin-positive GABAergic inhibitory interneurons, at least in the hippocampus and the temporal lobe [22].
Apart from SCN2A and SCN3A genes being affected by the presently detected deletion, GRB14 (exon 1 to intron 2–3), COBLL1 and SLC38A11 were also deleted in our patient. Thus, heterozygous deletion of these genes might also play a pivotal role in conferring ASD symptoms and could even be suggested to confer symptoms of Tourette syndrome in the present case: Loss of SLC38A11, a putative sodium-coupled neutral amino acid transporter [23] might enhance the effects of SCN2A and SCN3A deletion. The COBLL1 gene, encoding the Cordon-Bleu WH2 Repeat Protein Like 1 was suggested to be a negative regulator of apoptosis and associated with lower insulin resistance [24]. A deletion on chromosome 2q24.4 encompassing 47 genes including SCN2A2, SCN3A and COBLL1 was reported in a patient with severe epilepsy [14]. GRB14 encodes a growth factor receptor-binding protein acting as an inhibitor of intracellular signaling pathways regulating growth and metabolism [25]. One previous investigation reported a deletion on chromosome 2q comprising GRB14 and COBLL1 in a patient with autistic features, developmental delay, mental retardation, language impairment and dysmorphic features. In this case, additional repetitive hand movements have been described [2, 16]. Therefore, it is possible that deletions of GRB14 and COBLL1 may contribute to tic symptoms. An association of the above mentioned affected genes with Tourette syndrome proper, however, has not been reported in the presently available literature. From our point of view, the deletion therefore adequately explains the patient’s symptomatology. Furthermore, whole genome sequencing could provide further information on whether additional mutations/variants are present in the patient’s autistic brother contributing to the development of autistic symptoms in the patient.
Concerning treatment of SCN gene mutations, existing literature is really scarce. A causal treatment of deletions in chromosomal area 2q24.3 is not available. To date, a substitution of the missing gene products of the patient is not possible.
Promising studies reported a SCN2A mutation in a Chinese boy with infantile spasm responding to a modified Atkins Diet [26]. It is an “alternative” ketogenic diet with results demonstrating efficacy in the treatment of intractable seizures [27]. Evidence shows that ketones produced after a modified Atkins Diet could reduce neuron excitability by inhibiting glutamate transport and activating ATP-sensitive potassium channels [28]. Currently, there are no publications available investigating the effect of a ketogenic diet on Tic disorders or Tourette syndrome.
A review concluded that for the treatment of DS, which is often caused by loss-of-function mutations in SCN1A, valproate and benzodiazepines should be the first-line treatment but are often insufficient. Topiramate and levetiracetam, bromide and ketogenic diet provide efficacy as adjunctive therapy. Lamotrigine and carbamazepine should be avoided as they are supposed to lead to an increased occurrence of seizures [29]. Another promising drug is stiripentol showing efficacy in a combination with valproate and clobazam [29, 30]. Among patients with DS, a treatment with cannabidiol resulted in a reduction in seizure frequency compared to placebo, nevertheless higher rates of adverse events have been reported [31].
For the future, further research into targeted treatment options for SCN2A and SCN3A deletions is warranted. Previous studies primarily focused on patients with seizures and reported about medication with antiepileptic drugs or Atkins diets. In our case, the patient presented additional symptoms of Tourette syndrome, but no epilepsy. It remains to be elucidated whether the heterozygous loss of SCN2A and SCN3A or GRB14, COBLL1 and SLC38A11, respectively, might also contribute to the development of tics, which remains subject to investigation in large hypothesis-driven association studies.
Abbreviations
- ASD:
-
autism spectrum disorder
- ATP:
-
adenosine triphosphate
- COBL:
-
Cordon-Bleu
- DS:
-
Dravet-Syndrome
- DSM:
-
Diagnostic and Statistical Manual of Mental Disorders
- EEG:
-
electroencephalography
- FISH:
-
fluorescence in situ hybridization
- GABA:
-
gamma-aminobutyric acid
- GRB:
-
growth factor receptor-bound protein
- ICD:
-
International Statistical Classification of Diseases
- ISCN:
-
International System for Human Cytogenetic Nomenclature
- OGT:
-
Oxford Gene Technology
- OMIM:
-
Online Mendelian Inheritance in Man
- SCN:
-
sodium voltage-gated channel
- SLC:
-
solute carrier
References
Imbrici P, Camerino DC, Tricarico D. Major channels involved in neuropsychiatric disorders and therapeutic perspectives. Front Genet. 2013;4:76.
Celle ME, Cuoco C, Porta S, Gimelli G, Tassano E. Interstitial 2q24.3 deletion including SCN2A and SCN3A genes in a patient with autistic features, psychomotor delay, microcephaly and no history of seizures. Gene. 2013;532:294–6.
Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588(Pt 11):1841–8.
Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Råstam M, Sponheim E, et al. Genome-wide scan for autism susceptibility genes. Paris autism research international Sibpair study. Hum Mol Genet. 1999;8:805–12.
Weiss LA, Escayg A, Kearney JA, Trudeau M, MacDonald BT, Mori M, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8:186–94.
Bender AC, Morse RP, Scott RC, Holmes GL, Lenck-Santini P-P. SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cognitive outcome. Epilepsy Behav EB. 2012;23:177–86.
Le SV, Le PHT, Le TKV, Kieu Huynh TT, Hang Do TT. A mutation in GABRB3 associated with Dravet syndrome. Am J Med Genet A. 2017;173:2126–31.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41.
Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–7.
Kamiya K, Kaneda M, Sugawara T, Mazaki E, Okamura N, Montal M, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24:2690–8.
Liao Y, Anttonen A-K, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. 2010;75:1454–8.
Ogiwara I, Ito K, Sawaishi Y, Osaka H, Mazaki E, Inoue I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046–53.
Bartnik M, Chun-Hui Tsai A, Xia Z, Cheung SW, Stankiewicz P. Disruption of the SCN2A and SCN3A genes in a patient with mental retardation, neurobehavioral and psychiatric abnormalities, and a history of infantile seizures. Clin Genet. 2011;80:191–5.
Davidsson J, Collin A, Olsson ME, Lundgren J, Soller M. Deletion of the SCN gene cluster on 2q24.4 is associated with severe epilepsy: an array-based genotype-phenotype correlation and a comprehensive review of previously published cases. Epilepsy Res. 2008;81:69–79.
Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, et al. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci Lett. 2008;433:65–70.
Chen C-P, Lin S-P, Chern S-R, Chen Y-J, Tsai F-J, Wu P-C, et al. Array-CGH detection of a de novo 2.8 Mb deletion in 2q24.2-->q24.3 in a girl with autistic features and developmental delay. Eur J Med Genet. 2010;53:217–20.
Uddin LQ, Dajani DR, Voorhies W, Bednarz H, Kana RK. Progress and roadblocks in the search for brain-based biomarkers of autism and attention-deficit/hyperactivity disorder. Transl Psychiatry. 2017;7:e1218.
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46.
Gurney K, Prescott TJ, Wickens JR, Redgrave P. Computational models of the basal ganglia: from robots to membranes. Trends Neurosci. 2004;27:453–9.
Kalanithi PSA, Zheng W, Kataoka Y, DiFiglia M, Grantz H, Saper CB, et al. Altered parvalbumin-positive neuron distribution in basal ganglia of individuals with Tourette syndrome. Proc Natl Acad Sci U S A. 2005;102:13307–12.
Kataoka Y, Kalanithi PSA, Grantz H, Schwartz ML, Saper C, Leckman JF, et al. Decreased number of parvalbumin and cholinergic interneurons in the striatum of individuals with Tourette syndrome. J Comp Neurol. 2010;518:277–91.
Wang W, Takashima S, Segawa Y, Itoh M, Shi X, Hwang S-K, et al. The developmental changes of Na(v)1.1 and Na(v)1.2 expression in the human hippocampus and temporal lobe. Brain Res. 2011;1389:61–70.
Mackenzie B, Erickson JD. Sodium-coupled neutral amino acid (system N/a) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–95.
Han SH, Kim S-H, Kim H-J, Lee Y, Choi S-Y, Park G, et al. Cobll1 is linked to drug resistance and blastic transformation in chronic myeloid leukemia. Leukemia. 2017;31:1532–9.
Holt LJ, Siddle K. Grb10 and Grb14: enigmatic regulators of insulin action--and more? Biochem J. 2005;388(Pt 2):393–406.
Wong VCN, Fung CW, Kwong AKY. SCN2A mutation in a Chinese boy with infantile spasm - response to modified Atkins diet. Brain and Development. 2015;37:729–32.
Kossoff EH, Cervenka MC, Henry BJ, Haney CA, Turner Z. A decade of the modified Atkins diet (2003–2013): results, insights, and future directions. Epilepsy Behav EB. 2013;29:437–42.
Lutas A, Yellen G. The ketogenic diet: metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013;36:32–40.
Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol. 2011;53(Suppl 2):16–8.
Chiron C, Marchand MC, Tran A, Rey E, d’Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet Lond Engl. 2000;356:1638–42.
Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of Cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–20.
Acknowledgements
Not applicable.
Funding
There is no funding to report.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
KN, LTvE, KD, DE, SM and AR discussed, analysed and interpreted the patient data regarding clinical presentation, cytogenetic aspects and treatment options. KN, LTvE, KD, DE, SM and AR contributed to the manuscript. BG performed the CGH analysis, interpreted the data with respect to the cytogenetic aspects and created the fig. FS investigated and depicted the family history. KN and AR wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Authors’ information
Not applicable.
Ethics approval and consent to participate
Written informed consent was obtained from the parents of the patient for the case report.
Consent for publication
Written informed consent was obtained from the parents of the patient for publication of the case report.
Competing interests
Kathrin Nickel: no conflict of interest. Ludger Tebartz van Elst has received advisory boards, lectures, or travel grants within the last 3 years: Eli Lilly, Janssen-Cilag, Novartis, Shire, UCB, GSK, Servier, Janssen, and Cyberonics. Katharina Domschke: no conflict of interest. Birgitta Gläser: no conflict of interest. Friedrich Stock: no conflict of interest. Dominique Endres: no conflict of interest. Simon Maier: no conflict of interest. Andreas Riedel: no conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Nickel, K., Tebartz van Elst, L., Domschke, K. et al. Heterozygous deletion of SCN2A and SCN3A in a patient with autism spectrum disorder and Tourette syndrome: a case report. BMC Psychiatry 18, 248 (2018). https://doi.org/10.1186/s12888-018-1822-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-018-1822-8