
Abstract
Background
Tay-Sachs disease (TSD) is a sphingolipid storage disorder caused by mutations in the HEXA gene. To date, nearly 170 mutations of HEXA have been described, including only one 7.6 kb large deletion.
Methods
Multiplex Ligation-dependent Probe Amplification (MLPA) study was carried out in 5 unrelated patients for copy number changes where heterozygous and/or homozygous disease causing mutation/s could not be identified in the coding region by sequencing of HEXA gene.
Results
The study has identified the presence of a homozygous deletion of exon-2 and exon-3 in two patients, two patient showed compound heterozygosity with exon 1 deletion combined with missense mutation p.E462V and one patient was identified with duplication of exon-1 with novel variants c.1527-2A > T as a second allele.
Conclusion
This is the first report of deletion/duplication in HEXA gene providing a new insight into the molecular basis of TSD and use of MLPA assay for detecting large copy number changes in the HEXA gene.
Similar content being viewed by others

Background
Tay-Sachs disease (TSD) [MIM* 606869] is one of the common sphingolipid storage disorder in India [1]. It is a rare neurodegenerative lysosomal storage disorder (LSD) caused by a deficiency of ß-hexosaminidase-A (Hex-A) (HEXA; EC: 3.2.1.52) enzyme. It occurs due to the inability of Hex-A enzyme to cleave the terminal N-acetyl hexosamine residues from GM2 ganglioside due to a mutation in HEXA gene. As a result, GM2 ganglioside is accumulated in various tissues especially in neuronal cells instead of further metabolizing into GM3 gangliosides [2, 3]. The clinical phenotype varies widely with an acute infantile form of early onset leading to rapid neuroregression and early death to a progressive later onset form compatible with a longer survival [2].
The human HEXA gene is mapped on chromosomes 15q23-q24 with 35.56 kb spans, containing 14 exons [4]. As per HGMD (Human Gene Mutation Database), nearly 170 mutations have been reported so far in the gene that causes TSD; that include 130 single base substitutions, 29 small deletions, 6 small insertions, 2 indels and 1 large deletion of 7.6 kb (http://www.hgmd.cf.ac.uk/). Of these only 7.6 kb deletion is reported as a largest one in HEXA gene which covers 70% of infantile TSD cases in French Canadians [5].
Our earlier studies on Indian patients affected with TSD revealed various novel and known missense, nonsense, splice site mutation and frameshift mutations [6, 7]. In the present study, Multiplex Ligation-dependent Probe Amplification (MLPA) - based approach (MRC-Holland, P199-B) was used to investigate for the potential occurrence of large HEXA deletions/duplications in addition to common mutation(s) screening and bidirectional sequencing of HEXA gene.
Methods
The present study was carried out as a part of National Taskforce multicentric project of Indian Council of Medical Research (ICMR) and Department of Health Research (DHR), Government of India. The present study has been approved by the institutional ethics committee in accordance with the Helsinki declaration. A written informed consent was obtained from the parents before enrollment.
Patients
MLPA study was carried out in 5 enzymatically confirmed TSD patients for deletion/duplication analysis where disease causing mutation was not identified in the coding region of the gene and/or single disease causing allele was identified by common mutations screening and bi-directional sequencing of HEXA gene.
Multiplex ligation-dependent probe amplification (MLPA) analysis of HEXA gene
The genomic DNA was isolated from whole blood using salting out method [8]. MLPA analysis was carried out using P199-B2 HEXA P probe mix (MRC-Holland, Amsterdam, The Netherlands) in cases where Sanger sequencing failed to identify any pathological variant. The procedure was carried out according to the manufacturer’s recommendations using100 ng of genomic DNA. It was denatured at 98 °C for 5 min and hybridized overnight at 60 °C with the SALSA probe mix P199-B2 (HEXA gene, exons 1-14). Samples were then treated for ligation for 15 min at 54 °C. The reaction was stopped by incubation at 98 °C for 5 min. Finally, PCR amplification was carried out with the specific SALSA FAM PCR primers. Amplification products were run on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, USA). Copy number differences of various exons between test and control DNA samples were detected by analyzing the MLPA peak patterns.
Results
Molecular analysis was carried out in 75 TSD cases with deficiency of Hex-A and normal Total-Hex enzyme activity. Of these, 70 TSD patients have been identified with both coding mutations in HEXA gene while in 3 patients only one coding mutation was detected and in 2 patients no coding mutation was identified. Hence, MLPA study was carried out in these 5 unrelated patients to rule out copy number changes where heterozygous and/or homozygous disease causing variant could not be identified in the coding region by sequencing HEXA gene.
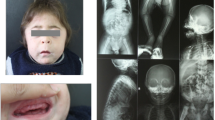
Consanguinity was present in 1/5 (20%) families. The mean age at presentation was 13.8 months (±2.48). All the cases were classified as infantile as they were presented with seizures, cherry red spot on the fundus, exaggerated startle, hypotonia, brisk deep tendon reflexes and regression of learned skill. The CT/MRI study of the brain was available in 3/5 cases and showed characteristic findings of a decrease in thalami and decreased attenuation of basal ganglia isodense with white matter, and one case had dysmyelination. A significant deficiency of Hex A activity was observed in the leukocytes of all five patients. The geographic/ethnic background, age at onset, age at last observation, enzyme activities and the genotypes identified are shown in Table 1.
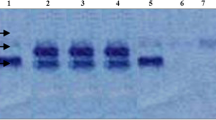
The MLPA analysis of HEXA gene showed the presence of homozygous deletion of exon-2 and exon-3 in two patients, two patients showed compound heterozygosity for exon 1 deletion and missense mutation p.E462V as a second allele and one patient was identified with duplication of exon-1 with novel splice site variant c.1527-2A > T as a second allele (Table 2 and Fig. 1). In Silico analysis of the novel variant was identified as disease causing by Mutation taster and NNsplice site 0.9 algorithm.

MLPA analysis of HEXA gene (a): homozygous deletion of exon 2 & 3; (b) & (c): heterozygous deletion of exon 1; (d): heterozygous duplication of exon 1
Discussion
The clinical appearance and neuroimaging features of infantile TSD seen in our patients were consistent with the defined phenotype. All patients presented with the severe infantile form of the disease irrespective of the genotype. The results of enzyme activity measurements (Hex-A expressed as a percentage of Total-Hex activity) varied from 0 to 0.2%. This is consistent with previous observations that infantile TSD patients have values ranging from 0 to 2% [9, 10].
During the course of the analysis we could not identify the second disease-causing allele in three patients and no variant was identified in two patients after sequencing the entire coding region of HEXA gene. Among the possible underlying reason for these findings, we suspected the presence of a deletion or duplication in the gene. Therefore using MLPA specific for HEXA gene we could identify two large deletions and one large duplication in an Indian TSD patients for the first time that include homozygous deletion of exon 2 & 3 in two cases, compound heterozygous deletion of exon-1 with second founder mutation p.E462V in two cases [6] and compound heterozygous duplication of exon-1 with second novel splice site variant c.1527-2A > T (Table 1). The carrier frequency of p.E462V mutation is ~ 1/500 which was earlier reported by Mistri et al. in 2012 [6]. In addition to this, recently the said variant was also reported only in one South Asian sample (http://gnomad.broadinstitute.org/variant/15-72638612-T-A). As has been known, the 7.6 kb deletion is the major mutation causing TSD in the French Canadian population; it removes part of intron-1, all of exon-1 and extends 2 kb upstream, encompassing the putative promoter region [4]. Although, this deletionwas never identified in our large cohort of Indian patient with TSD. Nonetheless, large deletion encompassing one or two exons or duplication of one exon are never reported and identified as a first disease causing variation in HEXA gene so far. Severity of phenotype in all five patients could be explained by the truncation of normal protein structure due to exon deletion/duplication in the gene.
This experimental approach of determination towards quantitative copy number variation in identifying large deletion and/or duplication is novel and reported here for the first time. The present study and earlier publications from our group [6, 7] also demonstrates that Indian TSD patients mainly portray infantile onset with severe phenotype irrespective of the genotype. None of our patients showed juvenile or late onset presentation. Though, it is highly likely that they are missed due to lack of awareness and failure of clinical identification as well. However, there are few mutations that have been identified in the late-onset phenotypes [11].
Conclusion
The present study demonstrates that large deletion and/or duplication in HEXA gene needs to beconsidered as the second tier approach in thegenomic sites where no variants are observed by conventional Sanger sequencing.
Abbreviations
- Hex-A:
-
β-hexosaminidase-A
- LSDs:
-
Lysosomal storage disorders
- MLPA:
-
Multiplex Ligation-dependent Probe Amplification
- TSD:
-
Tay-Sachs disease
References
Sheth J, Mistri M, Sheth F, Shah R, Bavdekar A, Godbole K, et al. Burden of lysosomal storage disorders in India: experience of 387 affected children from a single diagnostic facility. JIMD Rep. 2014;12:51–63.
Gravel RA, Kaback MM, Proia RL, Sandhoff K, Suzuki K. The GM2 gangliosidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. p. 3827–76.
Sonnino S, Chigorno V. Ganglioside molecular species containing C18- and C20-sphingosine in mammalian nervous tissues and neuronal cell cultures. Biochim Biophys Acta. 2000;1469:63–77.
Proia L, Neufeld EF. Synthesis of beta-hexosaminidase in cell-free translation and in intact fibroblasts: an insoluble precursor alpha chain in a rare form of Tay-Sachs disease. Proc Nat Acad Sci. 1982;79:6360–4.
Myerowitz R, Hogikyan ND. Different mutations in Ashkenazi Jewish and non-Jewish French Canadians with Tay-Sachs disease. Science. 1986;232(4758):1646–8.
Mistri M, Tamhankar P, Sheth F, Sanghavi D, Kondurkar P, Patil S, et al. Identification of novel mutations in HEXA gene in children affected with Tay Sachs disease from India. PLoS One. 2012;7(6):e39122.
Sheth J, Mistri M, Datar C, Kalane U, Patil S, Kamate M, et al. Expanding the spectrum of HEXA mutations in Indian patients with Tay-Sachs disease. Mol Genet Metab Rep. 2014;1:425–30.
Miller SA, Dykes DD. Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Kaback M, Desnick R. Pagon R, Bird T, Dolan C, Stephens K, editors. Hexosaminidase A Deficiency. Available from: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=tay-sachs (Accessed 17 Sept 2017)
Hou Y, Tse R, Mahuran DJ. The direct determination of the substrate specificity of the α-active site in heterodimeric β-hexosaminidase a. Biochemistry. 1996;35:3963–9.
Mahuran DJ. Biochemical consequences of mutations causing the GM2 gangliosidosis. Biochem Biophys Acta. 1999;1455:105–38.
Acknowledgements
We are grateful to the patients and their families who kindly agreed to participate in this study.
Funding
This work is supported by the Department of Health Research/Indian Council of Medical Research [grant no. BMS-54/2009], Government of India. Funding agency was not involved in the study design, specimen collection, analysis, interpretation of the data and preparation of the manuscript.
Availability of data and materials
Authors confirm that all the relevant data are included in the article.
Author information
Authors and Affiliations
Contributions
MM, JS were involved in the designing of the study, standardization of technical procedure, preparation of manuscript. MM was also involved in processing of sample for enzymes study and molecular analysis. JS will also act as a guarantor. MM, LM was involved in processing of samples for MLPA study. SM, MK, DS were involved in clinical information of all cases. FS has critically evaluated the manuscript. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Present case under submission has been approved by the institutional ethicscommittee [FRIGE’s Institute of Human Genetics] wide approval number FRIGE/IEC/5/2010 dated 7th March, 2010. This process is in accordance with the Helsinki declaration.
A written informed consent was obtained from the parents before enrollment[This was in accordance with the requirement of the institutional ethics committee].
Consent for publication
An informed consent for publication was also obtained from the parents of the subjects as they were minor [This was in accordance with the requirement of the institutional ethics committee].
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sheth, J., Mistri, M., Mahadevan, L. et al. Identification of deletion-duplication in HEXA gene in five children with Tay-Sachs disease from India. BMC Med Genet 19, 109 (2018). https://doi.org/10.1186/s12881-018-0632-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0632-7