
Abstract
Background
Asymptomatic nasopharyngeal carriage represents an important biological marker for monitoring pneumococcal serotype distribution and evaluating vaccine effects. Serotype determination by conventional method (Quellung reaction) is technically and financially challenging. On the contrary, PCR-based serotyping represents a simple, economic and promising alternative method.
Method
We designed a novel multiplex PCR assay for specific detection of the 30 classical colonizing S. pneumoniae serogroups/types. This multiplex assay is composed of 7 consecutive PCR reactions and was validated on a large and recent collection of Streptococcus pneumoniae isolated during a prospective study conducted in Belgium at the time of PCV7 adoption.
Results
The multiplex PCR assay allowed the typing of more than 94% of the isolates of a collection of pneumococci isolated from Belgian preschool attendees (n = 332). Seventy-five percent of the isolates were typed after 3 subsequent PCR reactions. Results were in agreement with the Quellung identification.
Conclusion
Our novel multiplex assay is an accurate and reliable method which can be used in place of the conventional method for S. pneumoniae carriage studies.
Similar content being viewed by others

Background
S. pneumoniae is a major cause of morbidity and mortality worldwide. The World Health Organisation estimates that 1.6 million people among which 0.7 to 1 million children under 5 die of pneumococcal diseases each year [1]. The nasopharynx of young children represents the ecological niche of S. pneumoniae as well as the reservoir for pneumococcal transmission within the community. Colonisation is also recognized as the first step of disease development [2]. S. pneumoniae major virulence factor is the capsular polysaccharide upon which S. pneumoniae classification is based. Today, 93 different serotypes (including the recently discovered 6D and 11E [3, 4]), belonging to 46 serogroups, have been described [5, 6]. Serotypes differ by geographical prevalence [7], attack rate [8], age distribution [7] tendency to cause outbreaks [9, 10] and propensity to acquire antimicrobial resistance genes [11–13]. The capsular polysaccharide is also the target of pneumococcal vaccine. The currently available protein conjugate vaccine, PCV7, protects against 7 clinically-relevant serotypes in young children (4, 6B, 9V, 14, 18C, 19F, 23F). Because of its coverage limitation, it has induced replacement of vaccine serotypes strains (VT) by non-vaccine serotypes strains (NVT) in colonisation and subsequently in disease [14, 15]. The long-term effects of a vaccine targeting only a restricted number of serotypes remain unknown. It is therefore crucial to keep ongoing surveillance of pneumococcal serotypes in invasive diseases [15, 16] and in carriage [17–19].
Serotype determination relies on the Quellung reaction. Binding of specific monoclonal antibodies with the capsule induces a capsular swelling visualised under the microscope. This technique remains the gold standard for pneumococcal serotyping but presents major drawbacks: high cost of antisera, technical expertise requirements and subjectivity in interpretation of results [20]. All these hindrances restrict S. pneumoniae serotyping to a few reference laboratories [21].
The capsular polysaccharides synthesis pathway is encoded by the cps (capsular polysaccharide synthesis) locus. Sequencing of the cps locus of the 93 known serotypes provided the molecular basis for PCR-based serotyping developments [22]. A first sequential multiplex PCR assay specific to invasive pneumococcal diseases (IPD) distribution in the USA was developed by Pai and colleagues [23]. This initial assay was modified based on IPD epidemiology encountered in different geographical areas (Brazil, Mozambique and Spain) [21, 24, 25]. It was also successfully applied for S. pneumoniae serotypes detection from nasopharyngeal (colonizing) and cerebro-spinal fluid (IPD) samples [21, 26–28] (Table 1). To conclude, most of the multiplex PCR assays developed so far are specific to IPD strains. Only two studies (in the USA and the Gambia) have used the IPD designed multiplex PCR in carriage studies [28, 29]. To optimize the rapid detection of carriage-associated serotypes, we developed a sequential multiplex PCR assay specific to colonizing pneumococci. This assay was experimentally tested on a collection of more than 300 pneumococci isolated in Belgium and representing carriage serotypes distribution in Europe. This new multiplex scheme successfully identified 95% of the isolates.
Methods
Study design
We carried out a prospective cohort study on healthy children (3-6 year-old) from 11 preschools of the Brussels area between September 2006 and May 2008, time of PCV7 introduction in Belgium [30]. Nasopharyngeal aspirates were performed three times during the school years on 333 children (mean age 4.2 years). A total of 830 samples were analysed for the presence of S. pneumoniae and 3 other potential pathogens (S. aureus, H. influenzae and M. catarrhalis). Sixty-nine percent of the children carried at least once S. pneumoniae. Number of colonies per sample was not determined. Only one colony of each bacterial species was included per sample.
Detailed information about the study was provided to parents and school staffs. The Ethics Committee of the Hôpital Universitaire des Enfants Reine Fabiola approved the study. Signed informed consents were obtained from parents.
Bacterial strains
A collection of 362 S. pneumoniae strains were isolated from healthy children [30]. S. pneumoniae isolates were identified by optochine-susceptibility test, bile solubility test and a positive lytA PCR as described previously [31].
S. pneumoniaetyping
S. pneumoniae isolates were serotyped by Quellung reaction performed by 2 different experimenters. Thirty-three different serogroups/types were identified [30]. Thirty isolates were assigned as non-typeable pneumococci (NTP). These 30 isolates were excluded from this study. The vast majority of these isolates did not show any amplification for the cpsA internal positive control (data not shown). A total of 332 isolates was therefore included in this study. Serotypes of these isolates were also determined using the original multiplex PCR assay described in Pai and colleagues [23]. Additional primers described in [25, 26] as well as primers specific to serotype 6C [32], 23A, 23B and 35A (newly designed by the CDC, (http://www.cdc.gov/ncidod/biotech/strep/pcr.htm)) were tested. All the primers used in this study have therefore been previously published (Table 2). Primers specific to the cpsA gene were used as an internal positive control. This gene is part of the cps locus and is highly conserved in pneumococci [23].
In case of non-concordant results, serotypes were double-checked using the Quellung reaction and the multiplex PCR assay. In case of non-concordance, the national reference laboratory confirmed the Quellung identification.
Molecular methods as DNA extraction, PCR and PCR product detection on 2% agarose gel were performed as described in [23]. Briefly, a bacterial suspension with a turbidity of MacFarland standard 1 was boiled at 100°C for 5 min and frozen at -20°C for 5 min. The PCR reactions were performed in 25 μl with 2.0 U of taq DNA polymerase (Promega Inc.), 5 μl of Promega green buffer 5X, 2.5 mM magnesium chloride, 200 μM of each DNTP (Promega Inc.), 2.5 μl of bacterial crude extract and primers with concentration as specified in Table 3. PCR reactions were performed using a Biometra T3000 thermocycler using the following conditions: 4 min at 94°C followed by 30 cycles composed of 45s at 94°C, 45s at 54°C and 150s at 65°C.
Results
In silico design of the multiplex PCR assay
A novel multiplex PCR assay was developed for specific detection of the 30 classical colonizing S. pneumoniae serogroups/types (Table 3). This multiplex assay is composed of 7 consecutive PCR reactions combining primers that have been already described in different studies [23, 25, 26] (Table 3). Serotypes included in the 7 PCR reactions were chosen upon their prevalence in carriage studies [7] to maximize serotypes determination in a limited number of PCR reactions. Primers were combined in each reaction to yield differences of more than 70 bp in PCR fragment size for non-ambiguous interpretation. Each PCR reaction includes a primer pair specific to the cpsA gene as internal positive control.
The specificity of the selected primer pairs was assessed in silico with the Primer-Blast software (http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) (default parameters with 'nr' database and 'Streptococcus pneumoniae taxid' species). Table 2 shows that 18 primer pairs are predicted to be specific to a given serotype (10A, 16F, 17F, 19A, 19F, 23A, 23B, 23F and 35B) or serogroup (1, 3, 4, 6, 14, 18, 22, 31 and 34) while the 12 others might present some cross-reactivity between different serotypes (7A/F, 9A/V, 11A/D/F, 15A/F, 15B/C and 24A/F) or serogroup (7C/B with 40; 12F with 12A/B, 44 and 46; 33F with 33A and 37; 35A with 35C and 42; 35F with 47F; 38 with 25A/F). Some serotypes are only occasionally associated with carriage and were therefore not included in the multiplex (e.g. serotypes 2, 5, 28, 29 and 46) [7].
Experimental validation of the multiplex PCR assay
The novel multiplex PCR assay was experimentally validated on a large and recent collection of S. pneumoniae isolated from Belgian preschool healthy children at the time of PCV7 adoption [30]. This collection included 332 typable isolates belonging to 31 serogroups/types assigned by Quellung reaction. Three serotypes (13, 28, 29) were not included in our PCR scheme. The PCR discriminates serotype 15A from serotypes 15B/C and serotype 35F from 35A. In total, 329 of the isolates (96%) belong to the 30 serotypes included in the PCR scheme. The 7 PCR reactions were tested on every isolate of this collection. During the experimental validation, we did not observe any primer inhibition and specific identification of each serotype was accurate (Figure 1).

Multiplex PCR reactions adapted to colonizing pneumococci. M means molecular size in bp.
The first PCR reaction (PCR-1) includes primers specific to serogroup/type 6, 23F, 11A and 23A. About one third of our collection was serotyped after PCR-1 (116/329 isolates, 35.25%) (Figure 2). Serogroup 6 primers cannot discriminate 6A and 6B isolates because of the high sequence similarity in the target gene (wciP). On the other hand, the newly discovered and emerging serotype 6C can be identified by PCR. We additionally tested the 60 serogroup 6 isolates using 6C primers (Primers described in Table 2, PCR condition as described in material and methods). Serotype 6C is recognized as an emerging serotype following PCV7 adoption [33, 34] and represents currently the predominant serogroup 6 serotype recovered in diseases in the USA [32]. No 6C isolates were present in our collection. The amplification of the internal control (cpsA) was present for 115 of the 116 strains identified by PCR-1. cpsA control amplification was missing for one 23A isolate (only the 23A specific amplicon of 723 bp was present). Twenty-two serotype 23A and 29 serotype 23F pneumococci are present in our collection. PCR-1 successfully identified 15 (68%) 23A and 22 (76%) 23F but no specific amplification was observed for 7 serotype 23A (32%) and 7 serotype 23F (24%). Subsequent testing of these strains with specific antisera at the National reference laboratory for pneumococci confirmed serotype 23A and 23F identification. The PCR identified accurately all other serotypes included in PCR-1 (6 and 11A). In total, PCR-1 successfully identified 116 (88%) of the 130 isolates belonging to serotypes 6, 23F, 11A and 23A. The experimental validation of the 4 primer pairs included in PCR-1 was 100% specific as no false positive results were obtained and no discrepancies with Quellung identification were observed. PCR-1 identified 37 serogroup 23 pneumococci but could not assign a serotype to 14 serogroup 23 strains.

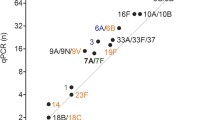
Serotype coverage of the Multiplex PCR. Number of pneumococcal isolates and cumulative frequencies for each serotype. C%, Cumulative Frequencies.
The second PCR reaction (PCR-2) is specific to serotypes 23B, 19F, 19A and 38. This reaction assigned a serotype to 69 isolates. After the 2 first reactions, more than half of the isolates were typed (185/329; 56.23%). cpsA internal control was missing for 9 out of 10 serotype 38 isolates but was present in every other isolate. In comparison to the Quellung serotyping, PCR-2 presented 97% sensitivity and 100% specificity. Only 2 out of 9 serotype 23B pneumococci (confirmed by the National reference laboratory) did not present any specific amplification.
The third reaction, PCR-3, detects serotypes 1, 3, 15B/C and 35B. This reaction allowed for serotype identification of 61 isolates. After 3 reactions, 74.77% of our isolates were serotyped and the data were in agreement with conventional serotyping (100% sensitivity and specificity).
PCR-4 (including serotypes 14, 34, 22F, 9A/V), PCR-5 (35A, 12F, 24A/F, 16F, 7A/F), PCR-6 (33F, 4, 35F, 10A, 31) and PCR-7 (7B/C, 15A, 17F, 18) allowed for the identification of 9%, 5%, 4% and 2% of the collection respectively (cumulative frequencies presented in Figure 2). The sensitivity and specificity were 100% for the last 4 PCR reactions.
In total, PCR-deduced results were concordant with conventional serotyping and allowed for the accurate identification of 313 out of 329 pneumococci of our collection (95.13%) (Figure 2).
Discussion
PCR schemes were developed to detect IPD associated serotypes distribution [21, 23–26]. Different authors have validated, adapted and improved the original multiplex PCR assay developed by Pai and colleagues [23]. These studies are summarized in table 1. The authors demonstrated the specificity of primers for IPD isolates in Africa [21], Asia [26], South-America [25] and Europe [24]. Moreover, PCR results were in agreement with conventional serotyping (91 to 100%) and a new PCR scheme was proposed for each location. The PCR multiplex assay was also used to detect carriage isolates in two studies [28, 29]. However, as serotypes associated with carriage and with diseases differ, the proposed PCR schemes were not optimal to rapidly serotype colonizing pneumococci. We thus designed a multiplex PCR scheme specific to carriage-associated serotypes. This assay can be used worldwide since serotype distribution of colonizing isolates is quite similar between different geographic locations. The most frequent serogroups are 6, 14, 19 and 23 [7]. Other colonizing serogroups frequently isolated from children are 3, 4, 9, 11, 13, 15, 18 and 33 [7].
The multiplex assay requires little technical skills and only basic PCR capabilities that are nowadays broadly accessible. Moreover, colonizing pneumococci represent a potent biological tool to monitor serotypes distribution and antimicrobial resistance level. Indeed, considering serotype prevalence in colonisation, rather than in disease, may be more accurate, faster and cheaper to evaluate pre- and post-vaccine product adoption [29]. The main disadvantage of PCR-based serotyping is the incapacity to discriminate closely-related serotypes such as 6A and 6B, 7F and 7A, 33F, 33A and 37, 35A, 35C and 42 (Table 2) but this accounts mainly for minor serotypes. Moreover, our in silico analysis of primer specificity showed that some primer pairs are predicted to present some cross-reactivity with other serogroups (e.g. primer pair for 7C/B, 12F, 33F, 35A, 35F and 38). This potential drawback could not be experimentally assessed in our collection as several serogroups were lacking in our collection. However, no false-positive result was obtained. As a consequence, when an amplicon of the proper size is obtained, isolates do not need to be tested with the following PCR reactions, as described previously [23].
The present scheme successfully typed 95.1% of our isolates belonging to serotypes included in the multiplex assay. This corresponds to 94.3% of the whole collection since primer pairs specific to 3 serotypes (13, 28 and 29, corresponding to three isolates) are not present in our assay. This validation confirms the flexibility of the multiplex PCR method because different primer pair combinations can be used. Moreover our method is rapid as 75% of our isolates were assigned a serotype after only 3 PCR reactions. For colonizing pneumococci, 2 studies evaluated the use of a PCR method to detect low colonisation rate and multiple colonizing serotypes from nasopharyngeal samples [28, 29]. This approach of direct serotype determination, without bacterial plating and isolation, is attractive but shows limitations such as incapacity to determinate antibiotics resistance profile or to apply complementary typing tools such as MLST.
Primers accurately identified the majority of serogroups/types included in our scheme but a lack of sensitivity of primers specific to serogroup 23 was observed. The sixteen strains for which no serotype could be assigned by PCR are belonging to serogroup 23 and represent 26.7% of the total serogroup 23 pneumococci isolated in our study (16/60). The Belgian serogroup 23 colonizing pneumococci present most likely sequence variability in the target gene (wzy for serotype 23A and 23F, wzx for serotype 23B) and are therefore not amplified by the current primer pairs. This demonstrates the need to validate the PCR-based method in different populations and locations to assess primers accuracy and specificity within a given serotype. Moreover, isolates that are non-typable by PCR should be monitored with Quellung technique to assess for the emergence of variants in pneumococcal serotypes.
In our collection, several pneumococci were assigned a serotype by multiplex PCR but did not present a cpsA-specific amplification (3%). This was already observed by Carvalho and colleagues at a rate of 1 to 2% [28]. This particularity is serotype dependent with serotype 38 and 25F generally negative for cpsA. We confirm this observation for colonizing serotype 38 (9 out of 10 isolates were cpsA negative). Variation within a serotype, incapacity to discriminate serotypes and occasional absence of the internal positive control represent the major weaknesses of the PCR-based method. These problems will likely be overcome in the near future with increasing sequencing of pneumococcal capsule locus. Sequence comparisons will enable the identification of conserved and specific sequences within a given serotype as well as the detection of conserved regions through 93 serotypes to design a reliable positive control. This will undoubtedly improve the PCR-based method.
Conclusions
To conclude, we have developed the first multiplex PCR assay designed for colonizing pneumococci. In the present study, we demonstrate that the PCR-based method is a simple, robust and economical method for serotype determination of colonizing pneumococci. We also demonstrate the accuracy of primers for carriage isolates in Belgium. This assay represents a good alternative to the Quellung reaction.
Abbreviations
- PCV7:
-
heptavalent pneumococcal conjugate vaccine
- VT:
-
vaccine types
- NVT:
-
non vaccine types
- NTP:
-
Non typeable pneumococci
- IPD:
-
invasive pneumococcal diseases
- bp:
-
base pair
- MLST:
-
Multi Locus Sequence Typing.
References
Pneumococcal conjugate vaccine for childhood immunization--WHO position paper. Wkly Epidemiol Rec. 2007, 82 (12): 93-104.
Bogaert D, De Groot R, Hermans PW: Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. 2004, 4 (3): 144-154. 10.1016/S1473-3099(04)00938-7.
Jin P, Kong F, Xiao M, Oftadeh S, Zhou F, Liu C, Russell F, Gilbert GL: First report of putative Streptococcus pneumoniae serotype 6D among nasopharyngeal isolates from Fijian children. J Infect Dis. 2009, 200 (9): 1375-1380. 10.1086/606118.
Calix JJ, Nahm MH: A new pneumococcal serotype, 11E, has a variably inactivated wcjE gene. J Infect Dis. 2010, 202 (1): 29-38. 10.1086/653123.
Henrichsen J: Six newly recognized types of Streptococcus pneumoniae. J Clin Microbiol. 1995, 33 (10): 2759-2762.
Park IH, Park S, Hollingshead SK, Nahm MH: Genetic basis for the new pneumococcal serotype, 6C. Infect Immun. 2007, 75 (9): 4482-4489. 10.1128/IAI.00510-07.
Hausdorff WP, Feikin DR, Klugman KP: Epidemiological differences among pneumococcal serotypes. Lancet Infect Dis. 2005, 5 (2): 83-93.
Sleeman KL, Griffiths D, Shackley F, Diggle L, Gupta S, Maiden MC, Moxon ER, Crook DW, Peto TE: Capsular serotype-specific attack rates and duration of carriage of Streptococcus pneumoniae in a population of children. J Infect Dis. 2006, 194 (5): 682-688. 10.1086/505710.
Le Hello S, Watson M, Levy M, Marcon S, Brown M, Yvon JF, Missotte I, Garin B: Invasive serotype 1 Streptococcus pneumoniae outbreaks in the South Pacific from 2000 to 2007. J Clin Microbiol. 2010, 48 (8): 2968-2971. 10.1128/JCM.01615-09.
Gupta A, Khaw FM, Stokle EL, George RC, Pebody R, Stansfield RE, Sheppard CL, Slack M, Gorton R, Spencer DA: Outbreak of Streptococcus pneumoniae serotype 1 pneumonia in a United Kingdom school. BMJ. 2008, 337: a2964-10.1136/bmj.a2964.
Richter SS, Heilmann KP, Coffman SL, Huynh HK, Brueggemann AB, Pfaller MA, Doern GV: The molecular epidemiology of penicillin-resistant Streptococcus pneumoniae in the United States, 1994-2000. Clin Infect Dis. 2002, 34 (3): 330-339. 10.1086/338065.
Feikin DR, Davis M, Nwanyanwu OC, Kazembe PN, Barat LM, Wasas A, Bloland PB, Ziba C, Capper T, Huebner RE, et al: Antibiotic resistance and serotype distribution of Streptococcus pneumoniae colonizing rural Malawian children. Pediatr Infect Dis J. 2003, 22 (6): 564-567.
Yagupsky P, Porat N, Fraser D, Prajgrod F, Merires M, McGee L, Klugman KP, Dagan R: Acquisition, carriage, and transmission of pneumococci with decreased antibiotic susceptibility in young children attending a day care facility in southern Israel. J Infect Dis. 1998, 177 (4): 1003-1012.
Huang SS, Hinrichsen VL, Stevenson AE, Rifas-Shiman SL, Kleinman K, Pelton SI, Lipsitch M, Hanage WP, Lee GM, Finkelstein JA: Continued impact of pneumococcal conjugate vaccine on carriage in young children. Pediatrics. 2009, 124 (1): e1-11. 10.1542/peds.2008-3099.
Hicks LA, Harrison LH, Flannery B, Hadler JL, Schaffner W, Craig AS, Jackson D, Thomas A, Beall B, Lynfield R, et al: Incidence of pneumococcal disease due to non-pneumococcal conjugate vaccine (PCV7) serotypes in the United States during the era of widespread PCV7 vaccination, 1998-2004. J Infect Dis. 2007, 196 (9): 1346-1354. 10.1086/521626.
Gonzalez BE, Hulten KG, Lamberth L, Kaplan SL, Mason EO: Streptococcus pneumoniae serogroups 15 and 33: an increasing cause of pneumococcal infections in children in the United States after the introduction of the pneumococcal 7-valent conjugate vaccine. Pediatr Infect Dis J. 2006, 25 (4): 301-305. 10.1097/01.inf.0000207484.52850.38.
Huang SS, Platt R, Rifas-Shiman SL, Pelton SI, Goldmann D, Finkelstein JA: Post-PCV7 changes in colonizing pneumococcal serotypes in 16 Massachusetts communities, 2001 and 2004. Pediatrics. 2005, 116 (3): e408-413. 10.1542/peds.2004-2338.
Huang SS, Finkelstein JA, Lipsitch M: Modeling community- and individual-level effects of child-care center attendance on pneumococcal carriage. Clin Infect Dis. 2005, 40 (9): 1215-1222. 10.1086/428580.
Sa-Leao R, Nunes S, Brito-Avo A, Frazao N, Simoes AS, Crisostomo MI, Paulo AC, Saldanha J, Santos-Sanches I, de Lencastre H: Changes in pneumococcal serotypes and antibiotypes carried by vaccinated and unvaccinated day-care centre attendees in Portugal, a country with widespread use of the seven-valent pneumococcal conjugate vaccine. Clin Microbiol Infect. 2009, 15 (11): 1002-1007. 10.1111/j.1469-0691.2009.02775.x.
Brito DA, Ramirez M, de Lencastre H: Serotyping Streptococcus pneumoniae by multiplex PCR. J Clin Microbiol. 2003, 41 (6): 2378-2384. 10.1128/JCM.41.6.2378-2384.2003.
Morais L, Carvalho Mda G, Roca A, Flannery B, Mandomando I, Soriano-Gabarro M, Sigauque B, Alonso P, Beall B: Sequential multiplex PCR for identifying pneumococcal capsular serotypes from South-Saharan African clinical isolates. J Med Microbiol. 2007, 56 (Pt 9): 1181-1184.
Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, Donohoe K, Harris D, Murphy L, Quail MA, et al: Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006, 2 (3): e31-10.1371/journal.pgen.0020031.
Pai R, Gertz RE, Beall B: Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J Clin Microbiol. 2006, 44 (1): 124-131. 10.1128/JCM.44.1.124-131.2006.
Iraurgui P, Torres MJ, Gandia A, Vazquez I, Cabrera EG, Obando I, Garnacho J, Aznar J: Modified sequential multiplex PCR for determining capsular serotypes of invasive pneumococci recovered from Seville. Clin Microbiol Infect.
Dias CA, Teixeira LM, Carvalho Mda G, Beall B: Sequential multiplex PCR for determining capsular serotypes of pneumococci recovered from Brazilian children. J Med Microbiol. 2007, 56 (Pt 9): 1185-1188.
Saha SK, Darmstadt GL, Baqui AH, Hossain B, Islam M, Foster D, Al-Emran H, Naheed A, Arifeen SE, Luby SP, et al: Identification of serotype in culture negative pneumococcal meningitis using sequential multiplex PCR: implication for surveillance and vaccine design. PLoS One. 2008, 3 (10): e3576-10.1371/journal.pone.0003576.
Njanpop Lafourcade BM, Sanou O, van der Linden M, Levina N, Karanfil M, Yaro S, Tamekloe TA, Mueller JE: Serotyping pneumococcal meningitis cases in the African meningitis belt by use of multiplex PCR with cerebrospinal fluid. J Clin Microbiol. 2010, 48 (2): 612-614. 10.1128/JCM.01402-09.
da Gloria Carvalho M, Pimenta FC, Jackson D, Roundtree A, Ahmad Y, Millar EV, O'Brien KL, Whitney CG, Cohen AL, Beall BW: Revisiting pneumococcal carriage by use of broth enrichment and PCR techniques for enhanced detection of carriage and serotypes. J Clin Microbiol. 2010, 48 (5): 1611-1618. 10.1128/JCM.02243-09.
Antonio M, Hakeem I, Sankareh K, Cheung YB, Adegbola RA: Evaluation of sequential multiplex PCR for direct detection of multiple serotypes of Streptococcus pneumoniae from nasopharyngeal secretions. J Med Microbiol. 2009, 58 (Pt 3): 296-302.
Jourdain S, Smeesters PR, Denis O, Dramaix M, Sputael V, Malaviolle X, Van Melderen L, Vergison A: Differences in nasopharyngeal bacterial carriage in preschool children from different socio-economic origins. Clin Microbiol Infect.
Sa-Leao R, Simoes AS, Nunes S, Sousa NG, Frazao N, de Lencastre H: Identification, prevalence and population structure of non-typable Streptococcus pneumoniae in carriage samples isolated from preschoolers attending day-care centres. Microbiology. 2006, 152 (Pt 2): 367-376.
Carvalho Mda G, Pimenta FC, Gertz RE, Joshi HH, Trujillo AA, Keys LE, Findley J, Moura IS, Park IH, Hollingshead SK, et al: PCR-based quantitation and clonal diversity of the current prevalent invasive serogroup 6 pneumococcal serotype, 6C, in the United States in 1999 and 2006 to 2007. J Clin Microbiol. 2009, 47 (3): 554-559. 10.1128/JCM.01919-08.
Whitney CG, Farley MM, Hadler J, Harrison LH, Bennett NM, Lynfield R, Reingold A, Cieslak PR, Pilishvili T, Jackson D, et al: Decline in invasive pneumococcal disease after the introduction of protein-polysaccharide conjugate vaccine. N Engl J Med. 2003, 348 (18): 1737-1746. 10.1056/NEJMoa022823.
Millar EV, O'Brien KL, Watt JP, Bronsdon MA, Dallas J, Whitney CG, Reid R, Santosham M: Effect of community-wide conjugate pneumococcal vaccine use in infancy on nasopharyngeal carriage through 3 years of age: a cross-sectional study in a high-risk population. Clin Infect Dis. 2006, 43 (1): 8-15. 10.1086/504802.
Azzari C, Moriondo M, Indolfi G, Massai C, Becciolini L, de Martino M, Resti M: Molecular detection methods and serotyping performed directly on clinical samples improve diagnostic sensitivity and reveal increased incidence of invasive disease by Streptococcus pneumoniae in Italian children. J Med Microbiol. 2008, 57 (Pt 10): 1205-1212.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2334/11/100/prepub
Acknowledgements
We thank Maria da Gloria Carvalho (CDC, Atlanta) for discussion and technical advices. We thank the Belgian Kids fund for financial support. SJ is supported by a grant from the Belgian Kids Fund. PRS is 'chargé de recherche' FNRS and has been supported by an ESPID Fellowship Award. This work was supported by the FNRS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
PRS has received a research grant from Pfizer.
JVe is member of a steering committee of a research project sponsored by Pfizer. JVe is heading the national reference laboratory for S. pneumoniae that received grants from Pfizer and GSK for serotyping isolates.
S J, PD, JV, LV: Nothing to declare
Authors' contributions
SJ and PRS conceived the study and wrote the paper with LV. SJ and PD performed multiplex PCR. SJ and JVa performed pneumococci serotyping. SJ, PRS, LV and Jve analysed the data. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jourdain, S., Drèze, PA., Vandeven, J. et al. Sequential multiplex PCR assay for determining capsular serotypes of colonizing S. pneumoniae . BMC Infect Dis 11, 100 (2011). https://doi.org/10.1186/1471-2334-11-100
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2334-11-100