
Abstract
Background
In this paper the interactions between paclitaxel, doxorubicin and the metabolic enzyme CYP3A4 are studied using computational models. The obtained results are compared with those of available clinical data sets. Analysis of the drug-enzyme interactions leads to a recommendation of an optimized paclitaxel-doxorubicin drug regime for chemotherapy treatment.
Methods
A saturable multi-compartment pharmacokinetic model for the multidrug treatment of cancer using paclitaxel and doxorubicin in a combination is developed. The model’s kinetic equations are then solved using standard numerical methods for solving systems of nonlinear differential equations. The parameters were adjusted by fitting to available clinical data. In addition, we studied the interaction of each drug with the metabolic enzyme CYP3A4 through blind docking simulations to demonstrate that these drugs compete for the same metabolic enzyme and to show their molecular mode of binding. This provides a molecular-level justification for the introduction of interaction terms in the kinetic model.
Results
Using docking simulations we compared the relative binding affinities for the metabolic enzyme of the two chemotherapy drugs. Since paclitaxel binds more strongly to CYP3A4 than doxorubicin, an explanation is given why doxorubicin has no apparent influence upon paclitaxel, while paclitaxel has a profound effect upon doxorubicin. Finally, we studied different time sequences of paclitaxel and doxorubicin concentrations and calculated their AUCs.
Conclusions
We have found excellent agreement between our model and available empirical clinical data for the drug combination studied here. To support the kinetic model at a molecular level, we built an atomistic three-dimensional model of the ligands interacting with the metabolic enzyme and elucidated the binding modes of paclitaxel and doxorubicin within CYP3A4. Blind docking simulations provided estimates of the corresponding binding energies. The paper is concluded with clinical implications for the administration of the two drugs in combination.
Similar content being viewed by others

Background
World Health Organization (WHO) statistics show that 12.5% of the deaths worldwide result from cancer related malignant neoplasms [1]. Thus, a critical need exists for effective treatments against these illnesses. Chemotherapy is the treatment of cancer using an anti-neoplastic drug or a combination of such drugs. There are more than one hundred distinct chemotherapy drugs approved for use in North America and Europe that can be divided into approximately ten categories. The first group consists of alkylating agents such as cisplatin. There are approximately twenty five different drugs in this group, which are genotoxic to the tumor cells. The second group contains plant alkaloids such as paclitaxel. There are approximately fifteen different drugs in this group and they inhibit the mitosis of the tumor cells. The remaining groups of chemotherapy agents are not relevant to the topic of this paper and hence will not be discussed. Combination cancer chemotherapies have shown to be more effective than mono-chemical therapy, with varying degrees of success. However, the question: “how can one best improve the outcome for a patient by changing the application frequency of a particular drug or by using a combination of drugs” has not been extensively studied in quantitative detail and it merits significant attention. The outcome of any therapy always involves both benefits and costs. The benefits can be measured in terms of the patient survival probability or the average time to recurrence. The cost can be assessed by the severity and frequency of side effects. Unfortunately, cancers are a set of very complex diseases for which finding cures has been extremely elusive. Part of the complexity describing cancer can be attributed to the unique and complicated signalling pathways leading to cancer initiation and progression at the level of individual cancer cells. Detailed maps of the signalling pathways have been found for a number of cancers and are catalogued in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). A typical KEGG pathway illustrates the molecular interactions and reaction networks for metabolism, various cellular processes, and human diseases (see [2]–[4]). For example, Figures 1 and 2 have been adapted from KEGG to illustrate two concrete examples how some of the available cancer chemotherapy drugs inhibit the cancer cell signaling pathways and how redundancies present in the signaling process complicate the situation.

Lung cancer signaling pathways. The red and blue dots illustrate the action of chemotherapy agents blocking the pathways.

Pancreatic cancer signaling pathways. The red and blue dots illustrate the action of chemotherapy agents blocking the pathways.
The accumulating knowledge about the molecular biology of cancer and novel delivery tools to specifically target aberrant proteins are opening up new therapeutic possibilities. One of the most common methods of increasing cure rates using chemotherapeutic agents is to administer combination chemotherapy which most often refers to the simultaneous administration of two or more medicinal compounds or modalities to treat a single disease. This approach in cancer treatment can be traced back to 1965 when James Holland, Emil Freireich, and Emil Frei hypothesized that cancer chemotherapy should follow the strategy of antibiotic therapy for tuberculosis and use combinations of drugs, each with a different mechanism of action. Cancer cells could conceivably mutate to become resistant to a single agent, but by using different drugs concurrently it would be more difficult for the tumor to develop resistance to the combination. Holland, Freireich, and Frei simultaneously administered an antifolate, a vinca alkaloid, 6-MP and Prednisone - referred to as the POMP regimen - and induced long-term remissions in children with ALL. This approach was extended to the lymphomas and other types of cancer. Currently, nearly all successful cancer chemotherapy regimens use this paradigm of multiple drugs given simultaneously. Some types of cancers, previously universally fatal, are now considered to be generally curable diseases as a result of this advance.
Current clinical trials in oncology commonly focus on three key aspects: (a) extending the scope of known drugs to new types of cancer, (b) testing new compounds, and (c) optimizing treatment by using combinations of known compounds. The latter aspect is of great interest and could benefit from a mathematical modelling approach aimed at achieving an optimized set of parameters for the dose, frequency and route of administration. This is, indeed, one of our main objectives behind the present work.
Two of the most widely used chemotherapy agents are paclitaxel and doxorubicin. Paclitaxel [5] is active against many human solid tumors related to breast cancer, ovarian cancer, non-small cell lung cancer, head and neck cancer, and advanced forms of Kaposi’s sarcoma [6]. Because it is poorly water-soluble, the current formulation for Taxol®; incorporates a 6 mg/ml solution in a solvent consisting of 50% polyoxyethylated castor oil (Cremophor®; EL-CrEL) and 50% dehydrated alcohol (USP). Paclitaxel is typically administered by IV infusion over one or three hours but has also been administered over six and twenty four hours. Because a patient may have an anaphylactic reaction to CrEL, alternative formulations of paclitaxel have been introduced, including BMS-184476, oral paclitaxel in polysorbate 80, and ABI-007, Genexol-PM [7] and Abraxane®; (nab-paclitaxel) [8]. Paclitaxel has a long residence time within the body and can stay trapped in cancer cells for over a week [9]. Paclitaxel is also highly bound to CrEL micelles, plasma proteins, platelets, and red blood cells [10].
DNA intercalators inhibit DNA polymerases and topoisomerases, resulting in the induction of apoptosis in tumor cells. DNA intercalating agents, such as amsacrine, actinomycin, mitoxantrone, and doxorubicin, have been employed as anticancer drugs and are in routine clinical use as chemotherapeutic agents [11]. It is well accepted that the antitumor activity of doxorubicin is caused by the formation of a cleavable complex of topoisomerase II, resulting in apoptosis [12],[13]. Doxorubicin is indicated in the treatment of a broad spectrum of solid tumors (e.g. breast, bladder, endometrium, thyroid, lung, ovary, stomach, and sarcomas of the bone) and in the treatment of lymphoma, as well as acute lymphoblastic and myeloblastic leukemias [14]. One of the most important and clinically relevant side effects of doxorubicin is the induction of cardiomyopathy [15]. A number of mechanisms have been proposed to explain this effect of doxorubicin, including oxidative stress [16], the induction of mitochondrial damage [17], and changes in gene expression in cardiac myocytes and muscle cells in general [18],[19].
Pharmacokinetics is the study of the absorption, distribution, metabolism and elimination (ADME) of drugs to, in, and from the body [20]. Pharmacological data usually consist of discrete values of the concentration of a drug in the plasma as a function of time. For drugs administered by direct intravenous (IV) infusion, a plot of these values generates a concentration-versus-time curve that rises during the infusion and then decreases after a maximum concentration value is reached. This decline may be relatively short or may last for several days, and it is mainly governed by the rate of elimination of the drug from the body. One of the key questions investigated is the functional dependence of the elimination curve and a single parameter that is often used to characterize the drug, namely its half-life. Another parameter of interest is the power law exponent of the elimination curve, which provides important information about the fate of the drug in the body and its efficacy. During clinical trials, the concentration-versus-time curves are used to determine optimum dosing regimens, potential toxicities, and drug-drug interactions.
The most common type of pharmacokinetic model is the compartmental model [20], in which a compartment is characterized by the number of drug molecules having the same probability of undergoing a set of chemical kinetic processes. The exchange of drug molecules between compartments is described by kinetic rate coefficients. Consider a one-compartment model post-infusion, such that
where k is the kinetic rate coefficient describing the elimination process. If the compartment is homogeneous and instantaneously-mixed (well-stirred), the kinetics are classical and
where k o is a constant. The resulting solution has an exponential tail,
Inhomogeneous compartments require a non-linear model and have been recently modeled using fractal kinetics [21],[22] with a time-dependent kinetic rate coefficient:
where η is a fractal exponent, which is related to the fractal dimension of the organ in which the process takes place. The resulting solution has a stretched exponential tail,
if η≠1. In the case η=1, the solution is a power-law relationship,
Saturable kinetics is usually modeled using Michaelis-Menten kinetics [23], where the differential equation describing a single compartment becomes
The quantity V max is the maximum rate of elimination, and K M is the amount of drug required to achieve half the maximum rate of the process (i.e. when X=K M then the rate of drug elimination is; . At very low dosage, the rate of elimination is given approximately by . Hence the model gives the standard expression for the rate of elimination at low dosage, and as the amount of drug increases the rate of elimination becomes a constant. The corresponding amount of drug versus time curve exhibits an initial linear segment (high dosage) followed by an exponential tail at low concentrations. The implicit solution to Equation 7, assuming constant parameters, is
A more complex single-compartment saturable model, which produces fractal effects, is given by the kinetic equation
which has the following exact solution, assuming constant parameters
The implicit solution in Equation 10 must be modified for two special cases. The first special case is when a=1, the first term must be replaced with . The second special case is when a−b=1, the second term must be replaced with .
Figure 3 graphically illustrates the solution given in Equation 10. In this figure; t o =1 hr, X o =10 μ mol/L, a=b, V max =18.80 μ mol/(L hr), and K M =5.50 (μ mol/L) are taken as an illustrative example.

Nonlinear saturable kinetics. The concentration-versus-time curve of the saturable kinetics example in the text for the solution given in Equation 10 with a=0.75 for the black curve, a=1 for the green curve, and a=2 for the red curve.
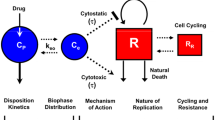
A single compartment model is clearly insufficient to capture the complexity of the pharmacokinetic and pharmacodynamic data exhibited by multi-drug chemotherapy. We believe that the simplest non-trivial model required for this purpose is a multi-compartment model in which the compartments are saturable due to the limitations on the number of enzymes and the molecular targets available for drug binding. The number of compartments considered for the modeling of the multi-drug pharmacokinetics of cancer chemotherapy agents should be at least four, namely: (1) the blood/plasma compartment, (2) the elimination organs compartment, (3) the healthy cells compartment, and (4) the tumor cells compartment. Compartment 1 represents the blood/plasma and is the mechanism for the transportation of the drug molecules throughout the body. Compartment 2 represents the liver and elimination organs, in which a portion of the drug is metabolized while the remaining drug is excreted from the body. Compartment 3 represents the healthy cells in the body and allows for a measure of the toxicity of the drugs. Compartment 4 represents the tumor cells. This model is clearly not as powerful as a physiologically-based approach that requires more detail as well as more model parameters describing its many compartments. However, we have decided to use the current four-compartment model to maintain simplicity in order to focus on the issue of drug interactions rather than the model’s physiological relevance and accuracy. We believe this model represents a compromise between simplicity and realistic representation of the PK system. This four-compartment model is illustrated in Figure 4.

Four-compartment pharmacokinetic model. The four compartments in the pharmacokinetic model presented in this paper: (1) the blood/plasma compartment, (2) the elimination organs compartment, (3) the healthy cells compartment, and (4) the tumor cells compartment.
The model presented here assumes well-stirred conditions, i.e. average drug concentrations in the various compartments are the only dependent variables. The mathematical model corresponding to Figure 4 is represented by a set of first-order (nonlinear) differential equations given by
where Xi,j is the concentration of drug i in compartment j, Ii,j is the rate of infusion divided by the volume of distribution of drug i in compartment j, compartment 0 (k=0 in the Equation 11 sum) is the environment external to the body, Ki,j,k is the kinetic rate coefficient for drug i moving from compartment j to compartment k, Γi,j,k is the saturation coefficient for drug i moving from compartment j to compartment k, and A(i,j,k) and B(i,j,k) are the fractal exponents for drug i moving from compartment j to compartment k.
The primary motivation for this paper is to quantitatively understand the combined action of paclitaxel and doxorubicin in view of the reported interactions when these drugs are administered simultaneously. Namely, it has been demonstrated that the combination of doxorubicin followed by infusion of paclitaxel has high antitumor activity in patients with metastatic breast cancer, but is strongly limited by the associated cardiac toxicity [24]. The question that arises is how these side effects can be reduced by optimal mode of administration of the two drugs. This requires careful analysis and model development. Both paclitaxel and doxorubicin are known to be substrates for specific metabolic enzymes. Such interactions with the metabolic enzymes raise the risk of alleviating their associated toxicity and inducing unpredictable adverse side effects due to metabolites. Given this clue, here we develop a predictive model of the pharmacokinetic and pharmacodynamic interactions between paclitaxel, doxorubicin and the metabolic enzymes in the body during multi-drug chemotherapy. Our pharmacokinetic model can be used in aiding the design of optimized dosage and scheduling of the two-drug combination.
In this paper, we discuss the results obtained at both levels of computational modeling: pharmacokinetics of the two drugs under investigation and molecular dynamics of their interactions with the metabolic enzyme.
Methods
Below is a short description of the pharmacokinetic model mentioned above. The model focuses on the interactions of paclitaxel and doxorubicin with a common metabolic enzyme for the two compounds. To gain further understanding of these interactions at the molecular level, we performed a blind docking simulation protocol, which was employed to predict the binding sites, modes of binding and binding energies of the two drugs to the CYP3A4 metabolic enzyme. This was intended to explain at a molecular level why these two drugs affect each other’s pharmacokinetic profiles. Below is a description of the modeling techniques used both for pharmacokinetic analysis and molecular dynamics simulations of the two drugs, paclitaxel and doxorubicin.
Pharmacokinetic modeling
The mathematical model corresponding to Figure 4 is Equation 11, which represents a system of nonlinear differential equations and in general, the kinetic rate coefficients do not have units of inverse time. Only in the special cases when all of the A’s are equal to one and the drugs have the same units in all the compartments, will the kinetic rate coefficients have units of inverse time. We defined I as the rate of infusion divided by the volume of distribution for the entire body, which is commonly known for most drugs from empirical data. In our case the volume of distribution is an adjustable parameter used to optimize fits to the available data sets. The volume of distribution for specific organs would be desirable but is very hard to determine, especially for human patients. We would require empirical data for the drug concentrations in several compartments in order to generate reliable parameter fits.
The parameters in Equation 11 are determined by minimizing the weighted percentage variance between the solution of Equation 11 and the clinical data. The weighted variance was used because some of the clinical concentration values were for a single patient while other clinical concentration values represented the mean value of the concentration for multiple patients. A percentage ratio was used because the concentrations varied over several orders of magnitude (approximately 0.01 to 10 micromoles per liter). The weighted percentage variance is defined as;
where Xi,j(t k ) is the solution of Equation 11 for the concentration of drug i in compartment j at time t k . is the concentration of drug i in compartment j at time t k from clinical study l. pl,i,j,k is the wieghting coefficient, which is the number of patients involved in study l used to measure and calculate the mean concentration . . In addition, the weighted variance is defined as;
The initial starting values for the parameters in Equation 11 were assumed to be for a linear system. i.e. the exponents were all set to one, the interaction parameters were set to zero, and the saturation parameters were set to zero. The clinical half-life for each of the studies was used to estimate the kinetic rate coefficients. The Xi,j(t k ) in Equation 12 were determined by solving the system of first order differential equations using standard numerical methods such as the Runge-Kutta Method. The maximum number of differential equations solved was fourteen for this system of drugs and their metabolites. The weighted percentage variance, VAR, can then be calculated for this specific set of parameters. The kinetic rate coefficients, the saturation parameters, and the exponents are then allowed to vary with the maximum number of parameters varying not exceeding the number of clinical data values for the concentrations minus one. The appropriate values for the parameters are obtained by finding the minimum value of VAR. The VAR can be minimized by varying these parameters using the Powell’s Method in Multidimensions.
An additional aspect over and above pharmacokinetic modeling that enhances our analysis is the molecular-level understanding of the nature of interactions between each of the two drugs and the metabolic enzyme CYP3A4. The latter effort requires an entirely different modeling methodology, namely molecular dynamics, which is briefly described below.
Molecular dynamics methodology
In order to determine if both doxorubicin and paclitaxel are substrates for the same metabolic enzyme, we used blind docking combined with molecular dynamics simulations to characterize the mode of action of the two drugs at the molecular level. We used the human microsomal cytochrome P450 3A4 crystal structure (PDB: entry 1TQN) [25]. The catalytic active site was well-characterized and included a HEM group. Prior to docking simulations, protonation states of the residues constituting the CYP3A4 including the HEM group were adjusted using the software PDB2PQR [25]. The protein structure was conformationally relaxed using the NAMD molecular dynamics software with constraints on the backbone atoms. The AMBER99SB force field [26] was used for protein parameterization, while the GAFF provided parameters for the HEM group.
To carry out the blind docking protocol, the entire surface of the CYP3A4 was divided into 90 focus docking regions. For each region, the center of mass of three solvent exposed neighboring atoms was used as the center of the docking box. The dimensions of the docking cube were 90×90×90 points with grid spacing of 0.03 nm. Clustering of the docking poses was performed using a 0.2 nm RMSD cut-off. To identify the most preferred binding locations, we combined all docking results and ranked them with the lowest binding energy of the largest docking cluster. As we are not only interested in the binding energies as indicators for adequate binding, we defined a hit as the docking run that includes at least 20% of the total population in its largest cluster. Finally, the solvent-exposed atoms that were used to construct the docking boxes have been used as markers for the binding locations.
All docking simulations were performed using AutoDock, version 4.0. Hydrogen atoms were added to all CYP3A4 and the two ligands followed by assigning their partial atomic charges using the Gasteiger-Marsili method. Atomic solvation parameters were assigned to the protein atoms using the AutoDock utility ADDSOL. A docking grid map with 90×90×90 points and grid point spacing of 0.03 nm has been calculated using AUTOGRID program. The grid box was centered on the active site. Rotatable bonds of each ligand were then automatically assigned using AUTOTORS utility of AutoDock. Docking was performed using the LGA method with an initial population of 400 random individuals, a maximum number of 10,000,000 energy evaluations, 100 trials, 50,000 maximum generations, a mutation rate of 0.02, a crossover rate of 0.80 and the requirement that only one individual can survive into the next generation.
Results and discussions
Pharmacokinetics of doxorubicin
The pharmacokinetics of a drug varies quite significantly between people and even in an individual person (pharmacokinetics will change with age and the health of the patient). Ideally, we would want a pharmacokinetic model with parameter values for each individual patient and the analysis would then be optimized for that specific patient; however, that is not practical. Therefore, we will calculate a set of parameters for a mean concentration versus time curve and obtain general qualitative features that will describe the people in this group as a whole.
The first stage is to determine the pharmacokinetics of doxorubicin from its concentration versus time clinical data and determine the parameters within the model in Equation 11 that fits this data without paclitaxel present. Equation 11 was fitted to several sets of experimental data for doxorubicin. The total number of data points that were digitized from the concentration-versus-time figures was 463. N p =1335, where N p is defined in Equation 12. The total number of parameters allowed to vary was sixty five, but only forty nine parameters were used. The minimization of VAR in Equation 12 produced the forty eight parameters in Table 1 for doxorubicin. In addition, the volume of distribution for doxorubicin was found to be VD,D O X=5.02 L/m2. The Table 1 numbers give a best fit to the doxorubicin data with the weighted percentage variance V A R=0.24. (The weighted variance is S=0.45 micromoles squared per liter squared.) The comparison of each set of experimental data with Equation 11 using the parameters given in Table 1 is given in Appendix Appendix A: Doxorubicin experimental data.
As an example, we have presented the data from Gianni et al., Moreira et al., and Benjamin et al. [27]-[29] in Figure 5, and plotted the model in Equation 11 for the dosage of 60 m g/m2 using the parameters in Table 1. The data from Gianni et al. [27] and Moreira et al. [28] stops at twenty four hours because that is when paclitaxel is introduced to the patients in those two studies. There are two sets of data from Benjamin et al. [29]; the pink data corresponds to chloroform being added to the extracted blood plasma and the purple data is for when an acid alcohol is added to the extracted blood plasma. In theory, these multiple patient studies should contain experimental data that overlaps along the same curve, but clearly that is not the case. The clinical data for doxorubicin is inconsistent from one study to the next with large variations in the results. The theoretical curve gives a best-fit of the data.

Comparison of theoretical and experimental doxorubicin results. The concentration-versus-time curve for doxorubicin. The black squares are the clinical data from Gianni et al. [27], the blue squares are the clinical data from Moreira et al. [28]. The purple and pink squares are the clinical data from Benjamin et al. [29]. The purple squares are for the acid alcohol data and the pink squares are for the chloroform data. The curve represents the solution of Equation 11 using the parameters in Table 1.
The metabolite, doxorubicinol, is produced from doxorubicin by the cytochrome P450 isoenzyme CYP3A4, and its pharmacokinetics can also be modeled using Equation 11. The total number of data points that were digitized from the concentration-versus-time figures was 126. N p =869, where N p is defined in Equation 12. The total number of parameters allowed to vary was fifty one, but only forty five parameters were used. The minimization of VAR in Equation 12 for doxorubicinol produced the forty parameters in Table 2 and five parameters in Equation 14. The weighted percentage variance is V A R=0.24 for doxorubicinol. (The weighted variance is S=0.0012 micromoles squared per liter squared for doxorubicinol). The combined weighted percentage variance is V A R=0.24 for doxorubicin and doxorubicinol. (The combined weighted variance is S=0.27 micromoles squared per liter squared for doxorubicin and doxorubicinol).
The rate at which doxorubicin is being converted into doxorubicinol was found to be:
The coefficient, 2.08, in Equation 14 represents the fraction of eliminated doxorubicin in compartment 2 being converted into doxorubicinol times the volume of distribution for doxorubicin divided by the volume of distribution for doxorubicinol. The comparison between Equation 11 using the parameters in Table 2 and each of the experimental set of data is given in Appendix Appendix A: Doxorubicin experimental data. As an example, we have presented the doxorubicin data and doxorubicinol data from Andersen et al. [30] and Greene et al. [31] in Figure 6.

Comparison of theoretical and experimental doxorubicinol results. The concentration-versus-time data for doxorubicin are represented by the squares and doxorubicinol data is represented by the triangles. The red data is from Andersen et al. [30] and the green data is from Greene et al. [31]. The curves are from Equation 11 using the parameters in Tables 1 and 2.
Note that some of the parameters for doxorubicin in Table 1 do not play a significant roll in fitting Equation 11 to the clinical data. One of A parameters in Table 1 is close to 1, and can be set to one. Two of the saturation parameters, Γ, are very small and two are zero. In addition, the 0.0002 in row 1,4 can be set to zero. These parameters can be ignored without losing much information. This would reduce the number of doxorubicin parameters to 42. In the case of doxorubicinol, all the parameters in Table 2 have an effect with one exception. The exponent A=1.03 could be set to 1 without a significant change to the results. Also, one of the three exponents in Equation 14, 0.98, is close to one and could be set to one as well. This reduces the number of doxorubicinol parameters to 43.
The best-fit parameters of doxorubicin and doxorubicinol in Tables 1 and 2 model all the data reasonably well, but not with enough accuracy for a detailed study of specific data sets because of the variations from one study to another as clearly shown in Figure 5. In order to study the interaction between doxorubicin and paclitaxel we need an accurate fit to the specific data set when there is no interaction between the two drugs. To acheive this, we begin with the best-fit parameters for all the data sets given in Tables 1 and 2 and perturb these parameters to improve the fit to the specific data set. We have adjusted and fitted the doxorubicin and doxorubicinol parameters to the clinical data obtained by Gianni et al. [27] and Moreira et al. [28] separately. The Gianni et al. [27] study had 10 doxorubicin data points and 8 doxorubicinol data points, when there was no paclitaxel present. The Moreira et al. [28] study had 10 doxorubicin data points and 10 doxorubicinol data points, when there was no paclitaxel present. N p =144, for Gianni et al. [27] and 560 for Moreira et al. [28],where N p is defined in Equation 12. We allowed all the parameters in Tables 1 and 2 to vary to determine, which parameters required the largest change. The result of fitting the theoretical model to the doxorubicin data of Gianni et al. [27] is shown in Table 3, and fitting the doxorubicinol parameters is shown in Table 4. The weighted percentage variance is V A R=1.7×10−3 and the weighted variance is S=3.3×10−4 micromoles squared per liter squared. The fit of the theoretical model to the data of Moreira et al. [28] is shown in Table 5 for doxorubicin, and the new doxorubicinol parameters are shown in Table 6. The weighted percentage variance is V A R=1.4×10−3 and the weighted variance is S=8.5×10−6 micromoles squared per liter squared. When relaxing the parameters to improve the fit with specific data, the parameters for the rate at which doxorubicin is being converted into doxorubicinol are changed in addition to the parameters in the tables. The new conversion rate for Gianni et al. [27] was found to be:
For Moreira et al. [28], the rate of conversion was found to be:
The parameters for both studies needed very little change. The result of these theoretical fits are shown in Figure 7 for Gianni et al. [27] and Figure 8 for Moreira et al. [28]. The agreement is very good as verified by the variance VAR and S defined in Equations 12 and 13, respectively.

Comparison of our theoretical results with the experimental results of Gianni et al. [[27]] for doxorubicin and doxorubicinol. The concentration-versus-time data for doxorubicin is represented by the squares and the doxorubicinol data is represented by the triangles. The data is from Gianni et al. [27], with the curve from Equation 11 using the parameters in Tables 3 and 4.

Comparison of theoretical and the experimental results of Moreira et al. [[28]] for doxorubicin and doxorubicinol. The concentration-versus-time data for doxorubicin is represented by the squares and the doxorubicinol data is represented by the triangles. The data is from Moreira et al. [28], with the curve from Equation 11 using the parameters in Tables 5 and 6.
Pharmacokinetics of paclitaxel
The pharmacokinetics of paclitaxel can be determined from its concentration-versus-time curves as well. We have digitized the data from several experimental data sets for paclitaxel and fitted to Equation 11. The number of clinical data points that were digitized from the concentration-versus-time figures and used to determine the parameters was 100. For these 100 data points N p =820, where N p is defined in Equation 12. The total number of parameters allowed to vary was sixty seven, but only fifty parameters were used. The minimization of VAR in Equation 12 produced the forty nine parameters for paclitaxel in Table 7 and the last parameter is the volume of distribution for paclitaxel, which was found to be VD,P T X=3.53 L/m2. The minimum weighted percentage variance was found to be V A R=0.035. The weighted variance was determined to be S=0.18 micromoles squared per liter squared. The results are shown in Figures 9 and 10. The experimental data in Figure 9 is from Gianni et al. [32]. The experimental data in Figure 10 is from Ohtsu et al. [33]. The comparison between the theoretical curve and rest of the experimental data is given in Appendix Appendix B: Paclitaxel experimental data.


Paclitaxel is metabolized by the cytochrome P450 isoenzymes CYP2C8 and CYP3A4. The primary metabolite, 6α-hydroxypaclitaxel (OHP), is formed by CYP2C8 while 3’-p-hydroxypaclitaxel and 6a,3’-p-dihydroxypaclitaxel are formed by CYP3A4. We can also model the metabolite(s) using Equation 11. We have digitized the experimental concentration-versus-time data and fitted the model in Equation 11 to the data. The total number of clinical data points that were digitized from the concentration-versus-time figures was 20. N p =173, where N p is defined in Equation 12. We fixed the exponents of the drug concentration to 1 because of the lack of data, and the coefficients were allowed to vary. There are twenty six coefficents, so those that had the least influence were set to zero. The minimization of VAR in Equation 12 produced the fifteen parameters for 6α-hydroxypaclitaxel in Table 8 and another three parameters in Equation 17. Equation 17 is the rate at which paclitaxel is being converted into 6α-hydroxypaclitaxel:
The coefficient 37.5 in Equation 17 represents the fraction of eliminated paclitaxel being converted into 6α-hydroxypaclitaxel times the volume of distribution for paclitaxel divided by the volume of distribution for 6α-hydroxypaclitaxel. The weighted percentage variance for 6α-hydroxypaclitaxel was calculated to be V A R=0.043 and the weighted variance was determined to be S=0.043 micromoles squared per liter squared without the paclitaxel. The combined weighted percentage variance for paclitaxel and its metabolite is V A R=0.036. The combined weighted variance is S=0.16 micromoles squared per liter squared for paclitaxel and its metabolite.
These models describe the pharmacokinetic effects of the drugs when there is no interaction between the drugs. The clinical data shows that they do influence each other’s metabolism. We intend to find out what the nature of their molecular interactions with the key metabolic enzyme are. In particular, we have hypothesized that both of the drugs compete for the same liver enzyme and the metabolic action of this enzyme, CYP3A4, can be saturated by one type of the drug molecules leaving the other unmetabolized. The drug with a higher binding free energy would have a greater probability of being metabolized. This is discussed in some detail below based on atomic level MD simulations.
The pharmacodynamic interaction of Doxorubicin and Paclitaxel with CYP3A4
The blind-docking simulations showed that both paclitaxel and doxorubicin bind to the surface of CYP3A4. Although the metabolic activity of CYP3A4 requires the binding of the substrate within the active site, the binding of the ligand should occur first on the surface of the enzyme. This initial binding would provide an entry point of the substrate to the active site. Based on this assumption, we investigated all possible binding locations for both doxorubicin and paclitaxel on the surface of CYP3A4. A blind docking analysis confirmed the binding of both compounds to CYP3A4. Figure 11 shows the binding locations of paclitaxel and doxorubicin on the surface of CYP3A4. The atoms of the protein are colored using the minimal value of their binding energy to the docked ligands. This procedure allowed us to identify and rank the most probable binding locations of the two drugs. The binding energies for paclitaxel ranged from −11 k c a l/m o l to −5 k c a l/m o l. On the other hand, doxorubicin bound to the surface of CYP3A4 with binding energies ranging from −8 k c a l/m o l to −4 k c a l/m o l. This analysis marked the possible entry points of the two ligands to the CYP3A4 active site. It is also worth mentioning that although the adopted blind docking procedure required considerable number of docking calculations, it has two advantages compared to covering the whole protein with a single docking box. First, the spacing between grid points used by AUTOGRID can be as accurate as a normal docking simulation. Second, there is no need to increase the number of energy evolutions or the population size to proportionally high values. This blind docking protocol was used earlier to successfully identify the binding location of laulimalide, a microtubule stabilizing agent, on the surface of tubulin [26].

Blind docking of paclitaxel and doxorubicin to CYP3A4. The blind docking illustration of paclitaxel (A) and doxorubicin (B) to CYP3A4. The red areas are the favorable binding regions and the blue areas are less favorable.
Figure 12 illustrates the binding modes of both paclitaxel and doxorubicin within the CYP3A4 active site. The protein possesses a relatively large substrate-binding cavity that accommodates the bulky paclitaxel substrate. The two compounds exhibited strong binding to the active site, although paclitaxel showed higher binding affinity. The binding energies of paclitaxel and doxorubicin were −12 k c a l/m o l and −10 k c a l/m o l, respectively. In the docked structures, paclitaxel was closer to the HEM group than doxorubicin. The two ligands formed hydrogen bonds with protein residues. For example, paclitaxel was hydrogen-bonded to residues Ile120, Leu211 and Arg372. Doxorubicin was attached to Phe108, Ile300 and Ala305. The large space available for both the ligands and protein residues allows for huge conformational changes that would accommodate the bound substrate. This correlates with similar available crystal structures of CYP3A4 bound to two distinct families of compounds [34].

Binding of paclitaxel and doxorubicin within CYP3A4. The binding modes of paclitaxel (A) and doxorubicin (B) within the active site of CYP3A4.
Based on the above simulations, it appears that both paclitaxel and doxorubicin strongly bind to the same metabolic enzyme, CYP3A4. Their binding locations are distinct, but conformational changes associated with the binding process may result in their competitive inhibition of each other. Furthermore, paclitaxel is predicted to bind significantly more strongly to CYP3A4 than doxorubicin giving a greater probability of being metabolized when administered simultaneously with doxorubicin. It is, therefore, reasonable to recommend the administration of doxorubicin after paclitaxel. This is further analyzed and quantified below.
The pharmacokinetic interaction between Doxorubicin and Paclitaxel
The metabolite, doxorubicinol, is produced from doxorubicin by the cytochrome P450 isoenzyme CYP3A4, while paclitaxel’s metabolites are produced by the cytochrome P450 isoenzymes CYP2C8 and CYP3A4. Paclitaxel’s primary metabolite is produced by CYP2C8 and hence doxorubicin is not expected to have a noticeable effect on the metabolism of paclitaxel. This is verified as illustrated in Figure 13. The three experimental studies shown in Figure 13 involve giving the patients 60 m g/m2 of doxorubicin over five minutes and then waiting for a period of time before giving the patients 200 m g/m2 of paclitaxel over a three-hour interval. The time for each set of paclitaxel data in Figure 13 was shifted so the time starts when the paclitaxel begins to be given to the patients. The green data corresponds to the administration of paclitaxel starting fifteen minutes after doxorubicin [27]. The black data corresponds to the administration of paclitaxel starting thirty minutes after doxorubicin [28]. The blue data corresponds to the administration of paclitaxel starting twenty four hours after doxorubicin [28]. The fact that the paclitaxel curves in Figure 13 do not change as a function of the time interval between the doxorubicin and paclitaxel administrations exemplifies that doxorubicin has no apparent effect upon the paclitaxel. The concentration-versus-time in Figure 13 also shows the theoretical curve. The theoretical curve is from Equation 11 using the parameters in Table 7 with no doxorubicin present. The three sets of clinical data and the theoretical curve agree remarkably well, which supports the assumption that doxorubicin has no influence on the paclitaxel.

The administration of paclitaxel after doxorubicin. The concentration-versus-time curve for 200 m g/m2 of paclitaxel given over a three-hour interval after the patient has recieved doxorubicin. The details about the data and the curve can be found in the discussion of the figure.
In contrast, we expect the presence of paclitaxel to have a strong influence on the metabolism of doxorubicin. As predicted in the previous subsection, paclitaxel will bind to CYP3A4 more strongly than doxorubicin and if the two drugs are simultaneously present, we expect paclitaxel to replace doxorubicin that is bound to CYP3A4. The doxorubicin and doxorubicinol parameters given in Tables 3 and 4 are for the best fit to the data from Gianni et al. [27], and the doxorubicin and doxorubicinol parameters in Tables 5 and 6 are for the best fit to the data from Moreira et al. [28]. The influence of paclitaxel on doxorubicin can be included in the model given in Equation 11 by modifying the rate coefficients, Ki,j,k given in these tables. The theoretical curve was fitted to the data by minimizing VAR in Equation 12. The number of data points in the Gianni et al. [27] study during the interaction of the drugs is 46 for doxorubicin (N p =568) and 48 for doxorubicinol (N p =494). The weighted percentage variance is V A R=0.069 and the weighted variance is S=1.6 micromoles squared per liter squared for doxorubicin. The weighted percentage variance is V A R=0.063 and the weighted variance is S=0.00010 micromoles squared per liter squared for doxorubicinol. The combined weighted percentage variance for all the drugs (145 data values and N p =1670) is V A R=0.068 and the combined weighted variance is S=1.1 micromoles squared per liter squared.
The total number of parameters used to minimize VAR was 10 for doxorubicin and 39 for doxorubicinol. These parameters are shown in Equation 18, and Tables 9 (doxorubicin) and 10 (doxorubicinol) for Gianni et al. [27]. In Table 10, the row 1,2 has a term with 0.00008 and the row 3,1 has a term with 0.00003. These eight parameters can be ignored without much lost of information. In addition, the rate at which doxorubicin is being converted into doxorubicinol was found to be
According to these parameters, the presence of the paclitaxel slows down the rate of elimination of the doxorubicin and doxorubicinol from the body. The paclitaxel also increases the kinetic rate at which doxoruciinol is moved from compartment 2 (elimination organs) to compartment 1 (blood/plasma) and reduces the kinetic rate for the reverse flow. The theoretical fit is shown in Figure 14.

Comparison of our theoretical results with the experimental results of Gianni et al. [[27]] for doxorubicin and doxorubicinol in the presence of paclitaxel. The squares represent the doxorubicin data, the triangles represent the doxorubicinol data, the circles represent the paclitaxel data, and the stars represent the 6α-hydroxypaclitaxel data. The concentration-versus-time results for different situations are shown in parts (A) and (B) of the figure, with the black curves being our theoretical predictions. 60 m g/m2 doxorubicin was given as a 0.083-hour bolus injection, followed by: (A) a 0.25-hour interval before 200 m g/m2 paclitaxel was infused over 3 hours. The green and blue data corresponds to two different sets of patients [27]. (B) a 24-hour interval before 150 m g/m2 paclitaxel was infused over 3 hours [27].
The doxorubicin and doxorubicinol parameters given in Tables 5 and 6 are for the best fit to the data from Moreira et al. [28]. The influence of paclitaxel on doxorubicin and doxorubicinol can be included in the model given in Equation 11 by modifying the rate coefficients, Ki,j,k given in these tables. The theoretical curve was fitted to the data by minimizing VAR in Equation 12. The number of data points in the Moreira et al. [28] study during the interaction of the drugs is 14 for doxorubicin (N p =392) and 16 for doxorubicinol (N p =448). The weighted percentage variance is V A R=0.0028 and the weighted variance is S=1.2×10−5 micromoles squared per liter squared for doxorubicin. The weighted percentage variance is V A R=0.028 and the weighted variance is S=0.00019 micromoles squared per liter squared for doxorubicinol. The combined weighted percentage variance for all the drugs (69 data values and N p =1932) is V A R=0.042 and the combined weighted variance is S=0.082 micromoles squared per liter squared.
The total number of parameters used to minimize VAR was 10 for doxorubicin and 24 for doxorubicinol. These parameters are given in Tables 11 (doxorubicin) and 12 (doxorubicinol) for Moreira et al. [28]. The rate of conversion of doxorubicin into doxorubicinol was found to be:
None of the parameters are insignificant. According to these parameters, the presence of the paclitaxel slows down the rate of elimination of the doxorubicin and doxorubicinol from the body. The paclitaxel repels the doxorubicin and doxorubicinol from the compartment. The paclitaxel in comparment 1 increases the rate of flow of doxorubicin and doxorubicinol into compartment 2 from 1 and slows down the flow from compartment 2 into 1. Similarly, the paclitaxel in comparment 2 increases the rate of flow of doxorubicin and doxorubicinol into compartment 1 from 2 and slows down the flow from compartment 1 into 2. The results for Moreira et al. [28] are shown in Figure 15.

Comparison of theoretical and the experimental results of Moreira et al. [[28]] for doxorubicin and doxorubicinol in the presence of paclitaxel. The squares represent the doxorubicin data, the triangles represent the doxorubicinol data, and the circles represent the paclitaxel data. The concentration-versus-time results for different situations are shown in parts (A) and (B) of the figure with the black curves being our theoretical predictions. 60 m g/m2 doxorubicin was given as a 0.083-hour bolus injection followed by: (A) 0.5-hour interval before 200 m g/m2 paclitaxel was infused over 3 hours. (B) 24-hour interval before 200 m g/m2 paclitaxel was infused over 3 hours [28].
The presence of paclitaxel within a compartment causes an increase in the rate at which doxorubicin flows from the compartment to the other compartment(s). In addition, the presence of paclitaxel slows the rate that doxorubicin is being eliminated from the body. The paclitaxel also causes the flow rate of the doxorubicinol to increase. The expulsion of the doxorubicin and doxorubicinol into the blood/plasma compartment is in agreement with what is observed experimentally with the spikes in the concentration-versus-time curves.
The amount of metabolite doxorubicinol being produced is also strongly affected by the presence of paclitaxel. We see from the expression for ID O L,2 that the fraction being metabolized into doxorubicinol is increased by the presence of paclitaxel; but the amount of doxorubicin in compartment 2 available to be converted into doxorubicinol is reduced as well as the amount being eliminated. Doxorubicinol is a highly cardiotoxic metabolite and the reduction of its production will reduce the chances of serious side effects [35].
The parameters in Tables 9 and 10 compared to Tables 11 and 12 are similar with a few differences. These differences do not change the overall qualitative behavior and produces similar results. For example, if doxorubicin is present and then paclitaxel is introduced to the body then both predict that the flow rate of doxorubicin into the elimination organs compartment will increase. Once the paclitaxel concentration in the elimination organ has increased to a sufficient level then the flow rate out into the blood plasma has increased more than the flow rate into the organ, which causes a spike in the concentration levels seen in the experimental data.
Our findings correlate well with the experimental data and provide computational and conceptual support for a possible binding of the two drugs to CYP3A4. The question that still remains is: what is the optimal time delay in the sequence of administration of the drugs? To answer this question, we calculate the AUC, which is defined as;
To investigate the time sequence we use the dosage introduced by Gianni et al. [27] and Moreira et al. [28]. In other words, 60 m g/m2 of doxorubicin is administered in a five-minute IV bolus and 150 m g/m2 or 200 m g/m2 of paclitaxel in a three-hour IV. The AUC is integrated over the period of infusion plus a one-week interval after the infusion has stopped instead of calculating the integral over all time. The portion of the curve neglected after a week has a very small contribution to the AUC, which does not change the results significantly.
The AUC for doxorubicin and doxorubicinol was calculated for the blood plasma compartment (1). In the case when doxorubicin is given first and paclitaxel is administered to the patient afterwards, the AUC of the doxorubicin is larger the closer in time the two are administered at. Unfortunately, the AUC of doxorubicinol is also very large the closer the two drugs are administered to each other reaching a maximum when both started at the same time. Therefore, we expect the toxicity of doxorubicin and doxorubicinol to be enhanced with this drug sequence. In the case when the order is switched and paclitaxel is given first there are two different situations. The first situation is when the doxorubicin is given during the first three hours while the paclitaxel is being administered. The AUC of the doxorubicin is a maximum during this interval. If the doxorubicin is given after the third hour the AUC becomes smaller the larger the interval between when the paclitaxel finishes and the doxorubicin is given. The AUC of the doxorubicinol is reduced by having the doxorubicin given after the paclitaxel. This reduces the toxicity of doxorubicinol while maximizing the effects of doxorubicin. Therefore, we maximize the effects of the combination if paclitaxel is given over three hours followed by doxorubicin given between 1 to 4 hours after the paclitaxel is started.
We conclude that doxorubicin should be administered optimally within four hours after the paclitaxel infusion is complete. These results suggest that the efficacy of doxorubicin can be increased by giving the patient paclitaxel prior to infusion of doxorubicin.
Conclusions
One of the major challenges in dose optimization is nonlinear behavior in one or more drug processes. In the current study, we investigated new ways to assess and quantify nonlinear pharmacokinetic behavior, with emphasis on their origins and therapeutic applications of the drug combination involving paclitaxel and doxorubicin. We have justified the development of a saturable compartment model with competing interactions between the two drugs by demonstrating through molecular dynamics simulations that they compete for the same metabolic enzyme with different binding affinities.
Molecular pathways are complex and the correlation of these pathways with malignancy is still an open issue, a complication that has led to systems biology approaches to cancer research [36]. While the system biology approach is promising, we focused on a well-understood and time-tested approach involving pharmacokinetic equations for a multi-compartment model leaving a more complex issue of multiple molecular pathways for future development.
Since no single drug is sufficiently efficacious to become a “silver bullet” cure for large patient populations, combinations of drugs addressing different molecular pathways appear to be capable of increasing patient survival. Optimization of combinations in terms of their sequences, schedules and dosages is a daunting task and needs a formal mathematical development. In the present paper we focused on a specific combination of two drugs and made the following two assumptions. First, saturation kinetics is in principle allowed in all compartments. Second, drug interactions are included to the extent of affecting kinetic rates of other drugs; specifically some drugs may either enhance or inhibit the transition kinetics of others depending on the built-in parameter values.
We have developed a relatively simple but robust four-compartment saturable kinetics model that has been shown to reproduce a number of empirical data sets very well. This is of practical importance in view of the toxic effects these drugs have on patients. In particular, it has been reported that doxorubicin is strongly cardio-toxic and doxorubicinol is ten times more cardio-toxic than doxorubicin [37]. Therefore, by keeping doxorubicin from being metabolized by liver enzymes, the drug will be more effective at killing the tumor as well as being less toxic to the body. This suggests that the optimal sequence is to give paclitaxel first and then almost immediately afterwards, give a dose of doxorubicin to the patient.
Appendix A: Doxorubicin experimental data
Equation 11 is the basis of our pharmacokinetic model and was fitted to several data sets that were digitized from concentration-versus-time curves. Some of the data represent mean values obtained from multiple patient sets, some of the data come from a representative patient out of a group, and some of the data describe a single patient. Due to this diversity of origin comparing different data sets is fraught with problems. However, our intention was to demonstrate the robustness of our model in fitting the data. In total, we have digitized 463 data points for doxorubicin. Multiplying the data points that represent mean values by the number of patients gave 1341 patient-data points. Similarly, for the metabolite doxorubicinol, we obtained 126 data points, which become 869 patient-data points when multiplied by the number of patients. The figures below show the individual data sets and our best fit.
The first set of data is from Andersen et al. [30]. This study involved 24 patients, each receiving 50 m g/m2 of doxorubicin in a 10-minute IV infusion. Figure 16 is a log-log plot of their doxorubicin and doxorubicinol data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2.

Doxorubicin and doxorubicinol from Andersen et al. Log-Log plot of doxorubicin and doxorubicinol. The doxorubicin data (squares) and the doxorubicinol data (triangles) are from Andersen et al. [30].
The second data set is from Greene et al. [31]. This study involved 10 patients, each receiving 75 m g/m2 of doxorubicin in a 15-minute IV infusion. Figure 17 is a log-log plot of their doxorubicin and doxorubicinol data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2.

Doxorubicin and doxorubicinol from Greene et al. Log-Log plot of doxorubicin and doxorubicinol. The doxorubicin data (squares) and the doxorubicinol data (triangles) are from Greene et al. [31].
The third data set is from Benjamin et al. [29]. This paper covered two separate studies. The first study involved 16 patients, each receiving 60 m g/m2 of doxorubicin in a 1 to 5 minute IV infusion. The second study involved 13 patients, each receiving 60 m g/m2 of doxorubicin in a 2 to 10 minute IV infusion. However, in the second study, doxorubicin and doxorubicinol was extracted from the plasma using chloroform: isopropyl alcohol (1:1, v/v). Figure 18 is a log-log plot of their doxorubicin and doxorubicinol data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2. The pink data correspond to the chloroform data and the purple was from the acid:alcohol extraction study.

Doxorubicin and doxorubicinol from Benjamin et al. Log-Log plot of doxorubicin and doxorubicinol. The doxorubicin data (squares) and the doxorubicinol data (triangles) are from Benjamin et al. [29].
The fourth data set is from Gianni et al. [27]. This study focused mainly on the interaction of doxorubicin with paclitaxel, but one of the published concentration-versus-time curves was for doxorubicin without any paclitaxel for the first 24 hours. This data involved 8 patients, each receiving 60 m g/m2 of doxorubicin in a 5-minute IV infusion. Figure 19 is a log-log plot of their doxorubicin and doxorubicinol data compared with the theoretical curve using Equation 11 with the parameter values given in the text.

Doxorubicin and doxorubicinol from Gianni et al. Log-Log plot of doxorubicin and doxorubicinol. The doxorubicin data (squares) and the doxorubicinol data (triangles) are from Gianni et al. [27].
The fifth data set is from Moreira et al. [28]. This study focused mainly on the interaction of doxorubicin with paclitaxel, but one of the published concentration-versus-time curves was for doxorubicin without any paclitaxel for the first 24 hours. This data involved 28 patients, each receiving 60 m g/m2 of doxorubicin in a 5-minute IV infusion. Figure 20 is a log-log plot of their doxorubicin and doxorubicinol data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2.

Doxorubicin and doxorubicinol from Moreira et al. Log-Log plot of doxorubicin and doxorubicinol. The doxorubicin data (squares) and the doxorubicinol data (triangles) are from Moreira et al. [28].
The sixth data set is from Di Fronzo et al. [38]. This study involved six patients, each receiving 0.5 m g/k g of doxorubicin. Figure 21 is a plot of their doxorubicin data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2.

Doxorubicin and doxorubicinol from Di Fronzo et al. Plot of doxorubicin data from Di Fronzo et al. compared to the theoretical curve [38].
The seventh, eighth, ninth, tenth and eleventh data sets are from Jacquet et al. [39]. This study involved 18 patients, each receiving 25 to 75 m g/m2 of doxorubicin in 5 to 15 minute IV infusion. The seventh study involved one patient and shows two courses of doxorubicin, each having a dose of 51.37 m g/m2 of doxorubicin in a 10-minute IV infusion. Figure 22 is a log-log plot of their doxorubicin data compared with the theoretical curve using Equation 11 with the parameter values given in Tables 1 and 2. The eighth study involves the same patient as the one in the seventh study and shows four courses of doxorubicin, each having a dose of 51.37 m g/m2 of doxorubicin in a 25-minute IV infusion, shown in Figure 23. The ninth study involves a single patient and shows four courses of doxorubicin, each having a dose of 51.12 m g/m2 of doxorubicin in a 15-minute IV infusion, which is illustrated in Figure 24. Figure 25 shows the tenth study, which involves the same patient as the ninth study and shows three courses of doxorubicin, each having a dose of 51.12 m g/m2 of doxorubicin in a 10-minute IV infusion. The eleventh study involves a patient who received three courses of doxorubicin, each having a dose of 45.9 m g/m2 of doxorubicin in a 15-minute IV infusion, which is shown in Figure 26.

10-minute IV of 51 . 37 m g / m2Doxorubicin. The squares represent the doxorubicin data from Jacquet et al. for 51.37 m g/m2 of doxorubicin during a 10-minute IV infusion [39].

25-minute IV of 51 . 37 m g / m2Doxorubicin. The squares represent the doxorubicin data from Jacquet et al. for 51.37 m g/m2 of doxorubicin during a 25-minute IV infusion [39].

15-minute IV of 51 . 12 m g / m2Doxorubicin. The squares represent the doxorubicin data from Jacquet et al. for 51.12 m g/m2 of doxorubicin during a 15-minute IV infusion [39].

10-minute IV of 51 . 12 m g / m2Doxorubicin. The squares represent the doxorubicin data from Jacquet et al. for 51.12 m g/m2 of doxorubicin during a 10-minute IV infusion [39].

15-minute IV or 45 . 9 m g / m2Doxorubicinol. The triangles represent the doxorubicinol data from Jacquet et al. for 45.9 m g/m2 of doxorubicin during a 15-minute IV infusion [39].
The twelfth data set is from Bressole et al. [40]. This study involved a single patient receiving 80 m g of doxorubicin during each course. We assumed that the body surface area was 1.6m2 and the infusion was over one hour. Figure 27 is a log-log plot of their doxorubicin data compared with the theoretical curve.

Doxorubicin from Bressole et al. The squares are the doxorubicin data from the Bressole et al. study [40]. The orange data is the third course and the red data is the sixth course.
The thirteenth data set is from Eksborg et al. [41]. Figure 28 is a log-log plot of their doxorubicin data compared with the theoretical curves using Equation 11 with the parameter values given in Tables 1 and 2. All the data are for individual patients. The blue data are for a dose of 33.8 m g/m2 in a 45-minute IV infusion. The pink data are for a dose of 49.3 m g/m2 in a 2-hour IV infusion. The red data are for a dose of 49.3 m g/m2 in a 4-hour IV infusion. The green data are for a dose of 30 m g/m2 in an 8-hour IV infusion. We assumed a 7.5-hour infusion for the theoretical curve. The black data are for a dose of 30 m g/m2 in a 16-hour IV infusion.

Doxorubicin from Eksborg et al. The doxorubicin data represented by the squares is from Eksborg et al. [41].
The fourteenth, fifteenth, sixteenth, seventeenth and eighteenth data sets are from Chan et al. [42]. All the theoretical curves are obtained from Equation 11 with the parameter values given in Tables 1 and 2. The fourteenth study shown in Figure 29 shows doxorubicin and doxorubicinol data for five patients, each receiving 45 m g/m2 of doxorubicin in a bolus IV. The fifteenth study shown in Figure 30 shows several courses given to a single patient, receiving 40 m g/m2 of doxorubicin in a bolus IV. Figure 31 shows four patients data, each receiving 30 m g/m2 of doxorubicin in a bolus IV. The gray data represents a few courses. Figure 32 shows data for four patients, each receiving 15 m g/m2 of doxorubicin in a bolus IV. Two courses of doxorubicin are shown for one of the patients. Figure 33 shows a patient receiving 10 m g/m2 of doxorubicin in a bolus IV.

Doxorubicin and doxorubicinol from Chan et al. A dosage of 45 m g/m2 of doxorubicin was administered to patients. The doxorubicin data represented by the squares and the doxorubicinol data represented by the triangles are from Chan et al. [42].

Doxorubicin from Chan et al. A dosage of 40 m g/m2 of doxorubicin was administered to patients. The doxorubicin data represented by the squares and the doxorubicinol data represented by the triangles are from Chan et al. [42].

Doxorubicin and doxorubicinol from Chan et al. A dosage of 30 m g/m2 of doxorubicin was administered to patients. The doxorubicin data represented by the squares and the doxorubicinol data represented by the triangles are from Chan et al. [42].

Doxorubicin and doxorubicinol from Chan et al. A dosage of 15 m g/m2 of doxorubicin was administered to patients. The doxorubicin data represented by the squares and the doxorubicinol data represented by the triangles are from Chan et al. [42].

Doxorubicin and doxorubicinol from Chan et al. A dosage of 10 m g/m2 of doxorubicin was administered to patients. The doxorubicin data represented by the squares and the doxorubicinol data represented by the triangles are from Chan et al. [42].
Appendix B: Paclitaxel experimental data
Equation 11 is the basis of our pharmacokinetic model and was fitted to several sets of paclitaxel data that were digitized from figures representing experimental concentration-versus-time curves. Some of the data represents mean values obtained from multiple patients, some of the data came from a representative patient out of a group, and some of the data describe a single patient. Due to this diversity of origin, we weighted each data set based on the number of patients it represented. We did not attempt to fit to a single set of data, but used all the data; our intention was to demonstrate the robustness of our model in fitting the data. In total, we have digitized 352 data points for paclitaxel. If we multiply the data points that represent mean values by the number of patients then we have 1104 patient-data points. Similarly, for the metabolite 6 α-hydroxylpaclitaxel, we obtained 20 data points, which is still 20 patient-data points when multiplied by the number of patients. The lack of data for the metabolite means we can use only 19 parameters and the fit is not as robust as it could have been with more experimental data. The figures below show the individual data sets and our best fits using Equation 11 with the parameter values given in Tables 7 and 8.
The first data set for paclitaxel is from Gianni et al. [32]. This study involved 30 patients; four patients received 135 m g/m2 during a three-hour IV infusion, three patients received 175 m g/m2 during a three-hour IV infusion, 15 patients received 225 m g/m2 during a three-hour IV infusion, four patients received 135 m g/m2 during a twenty four-hour IV infusion and 4 patients received 175 m g/m2 during a twenty four-hour IV infusion. Figure 34 is for a single representative patient for each dosage.

Paclitaxel and 6 α -hydroxylpaclitaxel from Gianni et al. The paclitaxel data represented by the circles and the 6 α-hydroxylpaclitaxel data represented by the diamonds are from Gianni et al. [32].
The second data set is from Monsarrat et al. [43]. This study involved a female patient receiving 135 m g/m2 during a three-hour IV infusion shown in Figure 35.

Paclitaxel and 6 α -hydroxylpaclitaxel from Monsarrat et al. The paclitaxel data represented by the circles and the 6 α-hydroxylpaclitaxel data represented by the diamonds are from Monsarrat et al. [43].
The third data set is from Mross et al. [44]. This study involved thirty patients; six patients received 150 m g/m2 during an one-hour IV infusion, six patients received 175 m g/m2 during an one-hour IV infusion, six patients received 200 m g/m2 during an one-hour IV infusion, six patients receiving 225 m g/m2 during an one-hour IV infusion and six patients receiving 250 m g/m2 during an one-hour IV infusion shown in Figure 36. The experimental data represent the median value of the concentrations of the six patients.

Paclitaxel from Mross et al. The paclitaxel data represented by the circles is from Mross et al. [44].
The fourth data set is from Brown et al. [45]. This study involved eighteen patients; seven patients receiving 175 m g/m2 during a six-hour IV infusion, six patients receiving 250 m g/m2 during a six-hour IV infusion, and five patients receiving 275 m g/m2 during a six-hour IV infusion, which is shown in Figure 37. Each set of experimental data in the figure corresponds to a single representative patient from the cohort.

Paclitaxel from Brown et al. The paclitaxel data represented by the circles is from Brown et al. [45].
The fifth data set is from Ohtsu et al. [33]. This study involved twenty two patients; three patients receiving 105 m g/m2 during a three-hour IV infusion, three patients receiving 180 m g/m2 during a three-hour IV infusion, seven patients receiving 240 m g/m2 during a three-hour IV infusion, three patients receiving 105 m g/m2 during a twenty four-hour IV infusion and six patients receiving 180 m g/m2 during a twenty four-hour IV infusion. The results are shown in Figure 38. The experimental data represent the mean values of the concentrations measured in the patients.

Paclitaxel from Ohtsu et al. The paclitaxel data represented by the circles is from Ohtsu et al. [33].
The sixth data set is from Longnecker et al. [46]. This study involved eight patients; two patients receiving 60 m g/m2 during an one-hour IV infusion, three patients receiving 170 m g/m2 during a six-hour IV infusion, and three patients receiving 265 m g/m2 during a six-hour IV infusion, which is shown in Figure 39. The experimental data represent individual patients from the study.

Paclitaxel from Longnecker et al. The paclitaxel data represented by the circles is from Longnecker et al. [46].
The seventh data set in Figure 40 is from Gelmon et al. [47]. This study involved five patients receiving 250 m g/m2 during a three-hour IV infusion. The experimental data represents a single patient out of the five patients in the study.

Paclitaxel from Gelmon et al. The paclitaxel data represented by the circles is from Gelmon et al. [47].
The eighth data set shown in Figure 41 is from Wiernik et al. [48]. This study involved twelve patients; four patients receiving 175 m g/m2 during a six-hour IV infusion, two patients receiving 200 m g/m2 during a six-hour IV infusion, three patients receiving 230 m g/m2 during a six-hour IV infusion and three patients receiving 275 m g/m2 during a six-hour IV infusion. The experimental data represent the mean values of the concentrations.

Paclitaxel from Wiernik et al. The paclitaxel data represented by the circles is from Wiernik et al. [48].
The ninth data set from Maier-Lenz et al. [49] is shown in Figure 42. This study involved thirty patients; four patients receiving 150 m g/m2 during an one-hour IV infusion, four patients receiving 175 m g/m2 during an one-hour IV infusion, thirteen patients receiving 200 m g/m2 during an one-hour IV infusion, eight patients receiving 225 m g/m2 during an one-hour IV infusion and three patients receiving 250 m g/m2 during an one-hour IV infusion. The experimental data represent the mean values of the concentrations.

Paclitaxel from Maier-Lenz et al. The paclitaxel data represented by the circles is from Maier-Lenz et al. [49].
Abbreviations
- ADME:
-
Absorption, distribution, metabolism and elimination
- ALA:
-
Alanine
- ALL:
-
Acute lymphoblastic leukaemia
- AMBER:
-
Assisted model building with energy Refinement
- ARG:
-
Arginine
- AUC:
-
Area under the curve
- CREL:
-
Cremphor EL
- CYP:
-
Cytochrome P450
- DNA:
-
Deoxyribonucleic acid
- DOL:
-
Doxorubicinol
- DOX:
-
Doxorubicin
- GAFF:
-
General amber force field
- HEM:
-
Hemoglobin
- ILE:
-
Isoleucine
- IV:
-
Intravenous
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LEU:
-
Leucine
- LGA:
-
Lamarckian genetic algorithm
- MD:
-
Molecular dynamics NAMD: Not (just) Another molecular dynamics program
- OHP:
-
6a-hydroxypaclitaxel
- PDB:
-
Protein data bank
- PHE:
-
Phenylalanine
- PK:
-
Pharmacokinetics
- PTX:
-
Paclitaxel
- RMSD:
-
Root mean square deviation
References
World Health Organization: WHO global report: Noncommunicable diseases country profiles 2011. Date: September, 2011 ISBN: 9789241502283 , [http://www.who.int/nmh/publications/ncd_profiles2011/en/]
KEGG PATHWAY Database - Metabolism, [http://www.genome.jp/kegg/pathway.html#metabolism] KEGG PATHWAY Database - Metabolism
KEGG PATHWAY Database - Cellular Processes, [http://www.genome.jp/kegg/pathway.html#cellular] KEGG PATHWAY Database - Cellular Processes
KEGG PATHWAY Database - Human Diseases, [http://www.genome.jp/kegg/pathway.html#disease] KEGG PATHWAY Database - Human Diseases
Rowinsky EK, Wright M, Monsarrat B, Lesser GJ, Donehower RC: Taxol: pharmacology, metabolism and clinical implications. Cancer Surv 1993, 17: 283–304.
Huizing MT, Misser VH, Pieters RC, ten Bokkel Huinink WW, Veenhof CH, Vermorken JB, Pinedo HM, Beijnen JH: Taxanes: a new class of antitumor agents. Cancer Invest 1995, 13: 381–404. 10.3109/07357909509031919
Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ: Phase i and pharmacokinetic study of genexol-pm, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res 2004, 10: 3708. 10.1158/1078-0432.CCR-03-0655
Robinson DM, Keating GM: Albumin-bound paclitaxel in metastatic breast cancer drugs. Drugs 2006, 66: 941. 10.2165/00003495-200666070-00007
Mori T, Kinoshita Y, Watanabe A, Yamaguchi T, Hosokawa K, Honjo K: Retention of paclitaxel in cancer cells for 1 week in vivo and in vitro. Cancer Chemother Pharmacol 2006, 58: 665–675. 10.1007/s00280-006-0209-6
Henningsson A, Karlsson MO, Vigano L, Gianni L, Verweij J, Sparreboom AA: Mechanism-based pharmacokinetic model for paclitaxel. J Clin Oncol 2001, 19: 4065.
Brana MF, Cacho M, Gradillas A, Pascual-Teresa B, Ramos A: Intercalators as anticancer drugs. Curr Pharm Des 2001, 7: 1745–1780. 10.2174/1381612013397113
Hickman JA, Beere HM, Wood AC, Waters CM, Parmar R: Mechanisms of cytotoxicity caused by antitumor drugs, 6th international congress of toxicology: toxicology from discovery and experimentation to the human perspective. Toxicol Lett 1992, 64–65: 553–561. 10.1016/0378-4274(92)90231-8
Kiechle FL, Zhang XB: Apoptosis: biochemical aspects and clinical implications. Clinica Chimica Acta 2002, 326: 27–45. 10.1016/S0009-8981(02)00297-8
Carter SK, Comis RL: Integration of chemotherapy into a combined modality approach for cancer treatment for pancreatic adenocarcinoma. Cancer Treat Rev 1975, 2: 193–214. 10.1016/S0305-7372(75)80003-X
Lenaz L, Page JA: Cardiotoxicity of adriamycin and related anthracyclines. Cancer Treat Rev 1975, 3: 111–120. 10.1016/S0305-7372(76)80018-7
Locker GY, Doroshow JH, Myers CE: Glutathione peroxidase - its role in adriamycin cardiotoxicity. In Proceedings of the American Association for Cancer Research: Volume 18. Philadelphia: American Association for Cancer Research; 1977:87.
Wallace KB: Doxorubicin-induced cardiac mitochondrionopathy. Pharmacol Toxicol 2003, 93: 105–115. 10.1034/j.1600-0773.2003.930301.x
Kurabayashi M, Jeyaseelan R, Kedes L: Doxorubicin represses the function of the myogenic helix-loop-helix transcription factor myod - involvement of id gene induction. J Biol Chem 1994, 269: 6031–6039.
Boucek RJ, Miracle A, Anderson M, Engelman R, Atkinson J, Dodd DA: Persistent effects of doxorubicin on cardiac gene expression. J Mol Cell Cardiol 1999, 31: 1435–1446. 10.1006/jmcc.1999.0972
Gibaldi M, Perrier D: Pharmacokinetics. 2nd edition, New York: M. Dekker; 1982. Gibaldi M, Perrier D: Pharmacokinetics. 2nd edition, New York: M. Dekker; 1982.
Macheras P: A fractal approach to heterogeneous drug distribution: calcium pharmacokinetics. Pharm Res 1996, 13: 663–670. 10.1023/A:1016031129053
Fuite J, Marsh RE, Tuszynski JA: Fractal pharmacokinetics of the drug mibefradil in the liver. Phys Rev E 2002, 66: 021904. 10.1103/PhysRevE.66.021904
Marsh RE, Tuszynski JA, Sawyer MB, Vos KJE: Emergence of power laws in the pharmacokientics of paclitaxel due to competing saturable processes. J Pharmacy Pharmaceutical Sci 2008, 11: 77–96.
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C: Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65: 712–25. 10.1002/prot.21123
Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF: The structure of human microsomal cytochrome p450 3a4 determined by x-ray crystallography to 2.05-a resolution. J Biol Chem 2004, 279: 38091–38094. 10.1074/jbc.C400293200
Bennet MJ, Barakat K, Huzil JT, Tuszynski JA, Schriemer DC: Discovery and characterization of the laulimalide-microtubule binding mode by mass shift perturbation mapping. Chem Biol 2010, 17: 725–734. 10.1016/j.chembiol.2010.05.019
Gianni L, Vigano L, Locatelli A, Capri G, Giani A, Tarenzi E, Bonadonna G: Human pharmacokinetic characterization and in vitro study of the interaction between doxorubicin and paclitaxel in patients with breast cancer. J Clin Oncol 1997, 15: 1906–1915.
Moreira A, Lobato R, Morais J, Silva S, Ribeiro J, Figueira A, Vale D, Sousa C, Araujo F, Fernandes A, Oliveira J, Passos-Coelho JL: Influence of the interval between the administration of doxorubicin and paclitaxel on the pharmacokinetics of these drugs in patients with locally advanced breast cancer. Cancer Chemotherapy Pharmacol 2001, 48: 333–337. 10.1007/s002800100297
Benjamin RS, Riggs CE, Bachur NR: Influence of the interval between the administration of doxorubicin and paclitaxel on the pharmacokinetics of these drugs in patients with locally advanced breast cancer. Cancer Chemotherapy Pharmacol 2001, 48: 333–337. 10.1007/s002800100297
Andersen A, Holte H, Slordal L: Pharmacokinetics and metabolism of doxorubicin after short-term infusions in lymphoma patients. Cancer Chemotherapy Pharmacol 1999, 44: 422–426. 10.1007/s002800050999
Greene RF, Collins JM, Jenkins JF, Speyer JL, Myers CE: Plasma pharmacokinetics of adriamycin and adriamycinol: Implications for the design of in vitro experiments and treatment protocols. Cancer Res 1983, 43: 3417–3421.
Gianni L, Kearns CM, Giani A, Capri G, Vigano L, Locatelli A, Bonadonna G, Egorin MJ: Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol 1995, 13: 180–190.
Ohtsu T, Sasaki Y, Tamura T, Miyata Y, Nakanomyo H, Nishiwaki Y, Saijo N: Clinical pharmacokinetics and pharmacodynamics of paclitaxel: a 3-hour infusion versus a 24-hour infusion. Clin Cancer Res 1995, 1: 599–606.
Ekroos M, Sjogren T: Structural basis for ligand promiscuity in cytochrome p450 3a4. Proc Natl Acad Sci U S A 2006, 103: 13682–13687. 10.1073/pnas.0603236103
Boucek RJ, Olson RD, Brenner DE, Ogunbunmi EM, Inui M, Fleischer S: The major metabolite of doxorubicin is a potent inhibitor of membrane-associated ion pumps: A correlative study of cardiac muscle with isolated membrane fractions. J Biol Chem 1987, 262: 15851–15856.
Hornberg JJ, Bruggeman FJ, Westerhoff HV, Lankelma J: Cancer: a systems biology disease. Biosystems 2006, 83: 81–90. 10.1016/j.biosystems.2005.05.014
Mross K, Niemann B, Massing U, Drevs J, Unger C, Bhamra R, Swenson C: Pharmacokinetics of liposomal doxorubicin (tlc-d99; myocet) in patients with solid tumors: an open-label, single-dose study. Cancer Chemother Pharmacol 2004, 54: 514–524. 10.1007/s00280-004-0825-y
Di Fronzo G, Lenaz L, Bonadonna G: Distribution and excretion of adriamycin in man. Biomedicine 1973, 19: 169–171.
Jacquet JM, Bressolle F, Galtier M, Bourrier M, Donadio D, Jourdan J, Rossi JF: Doxorubicin and doxorubicinol: intra- and inter-individual variations of pharmacokinetic parameters. Cancer Chemother Pharmacol 1990, 27: 219–225. 10.1007/BF00685716
Bressolle F, Ray P, Jacquet JM, Bres J, Galitier M, Donadio D, Jourdan J, Rossi JF: Bayesian estimation of doxorubicin pharmacokinetic parameters. Cancer Chemother Pharmacol 1991, 29: 53–60. 10.1007/BF00686336
Eksborg S, Strandler HS, Edsmyr F, Naslund I, Tahvanainen P: Pharmacokinetic study of iv infusions of adriamycin. Eur J Pharmacol 1985, 28: 205–212. 10.1007/BF00609693
Chan KK, Chlebowski RT, Tong M, Chen HSG, Gross JF, Bateman JR: Clinical pharmacokinetics of adriamycin in hepatoma patients with cirrhosis. Cancer Res 1980, 40: 1263–1268.
Monsarrat B, Chatelut E, Royer I, Alvineirie P, Dubois J, Dezeuse A, Roche H, Cros S, Wright M, Canal P: Modification of paclitaxel metabolism in a cancer patient by induction of cytochrome p450 3a4. Drug Metab Dispos 1998, 26: 229–233.
Mross K, Hollander N, Hauns B, Schumacher M, Maier-Lenz H: The pharmacokinetics of a 1-h paclitaxel infusion. Cancer Chemother Pharmacol 2000, 45: 463–470. 10.1007/s002800051020
Brown T, Havlin K, Weiss G, Cagnola J, Koeller J, Kuhn J, Rizzo J, Craig J, Phillips J, Von Hoff D: A phase 1 trial of taxol given by a 6-hour intravenous infusion. J Clin Oncol 1991, 9: 1261–1267.
Longnecker SM, Donehower RC, Cates AE, Chen TL, Brundrett RB, Grochow LB, Ettinger DS, Colvin M: High-performance liquid chromatography assay for taxol in human plasma and urine and pharmacokinetics in a phase i trial. Cancer Treat Rep 1987, 71: 53–59.
Gelmon K, Eisenhauer E, Bryce C, Tolcher A, Mayer L, Tomlinson E, Zee B, Blackstein M, Tomiak E, Yau J, Batist G, Fisher B, Iglesias J: Randomized phase ii study of high-dose paclitaxel with or without amifostine in patients with metastatic breast cancer. J Clin Oncol 1999, 17: 3038–3047.
Wiernik PH, Schwartz EL, Strauman JJ, Dutcher JP, Lipton RB, Paietta E: Phase I clinical and pharmacokinetic study of taxol. Cancer Res 1987, 47: 2486–2493.
Maier-Lenz H, Hauns B, Haering B, Koetting J, Mross K, Unger C, Bauknecht T, duBois A, Meerpohl HG, Hollaender N, Diergarten K: Phase i study of paclitaxel administration as a 1-hr infusion: Toxicity and pharmacokinetics. Semin Oncol 1997, 24: 19–161919.
Acknowledgements
J.A.T. acknowledges funding support for this project from NSERC, the Allard Foundation, the Canadian Breast Cancer Foundation and the Alberta Advanced Education and Technology. K.J.E.V. would like to thank J.A.T., the Cross Cancer Institute and the University of Alberta for their hospitality.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KJEV developed the model in Equation 11, developed the numerical code for fitting the model to the experimental data, was involved in running the numerical code, and was involved in writing the manuscript. AGM was involved in collecting experimental data from the literature for both paclitaxel and doxorubicin, digitalizing the experimental concentration-versus-time data, and fitting the theoretical model to the experimental data. MGT was involved in collecting experimental data from the literature for both paclitaxel and doxorubicin, digitalizing the experimental concentration-versus-time data, and fitting the theoretical model to the experimental data. LF was involved in collecting experimental data from the literature for doxorubicin, digitalizing the experimental concentration-versus-time data, and fitting the theoretical model to the experimental data. KB was involved in all aspects of the molecular dynamics simulations. JAT was involved with the development of the model in Equation 11, development of the molecular dynamics simulations, and was involved in writing the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Vos, K.J., Martin, A.G., Trimboli, M.G. et al. A multi-compartment pharmacokinetic model of the interaction between paclitaxel and doxorubicin. EPJ Nonlinear Biomed Phys 2, 13 (2014). https://doi.org/10.1140/epjnbp/s40366-014-0013-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjnbp/s40366-014-0013-x