
Abstract
Increasing the accuracy of pathogen identification and reducing the duration of analysis remain relevant for modern molecular diagnostics up to this day. In laboratory and clinical practice, detection of pathogens mostly relies on methods of nucleic acid amplification, among which the polymerase chain reaction (PCR) is considered the “gold standard.” Nevertheless, in some cases, isothermal amplification methods act as an alternative to PCR diagnostics. Upon more than thirty years of the development of isothermal DNA synthesis, the appearance of loop-mediated isothermal amplification (LAMP) has enabled new directions of in-field diagnostics of bacterial and viral infections. This review examines the key characteristics of the LAMP method and corresponding features in practice. We discuss the structure of LAMP amplicons with single-stranded loops, which have the sites for primer annealing under isothermal conditions. The latest achievements in the modification of the LAMP method are analyzed, which allow considering it as a unique platform for creating the next-generation diagnostic assays.
Similar content being viewed by others

INTRODUCTION
Nucleic acid amplification methods have found wide application in various fields of molecular biology and become an indispensable foundation for genetic engineering and a valuable source of information on the causes of infectious and hereditary diseases. The polymerase chain reaction (PCR) method is still in great demand in solving a wide range of research problems. Despite the important role of this method in healthcare, when creating mobile diagnostic methods, a group of various methods based on isothermal amplification technology has become an attractive alternative to PCR [1, 2].
With isothermal amplification, in contrast to PCR, there is no need for temperature cycles for denaturation of nucleic acid and annealing and elongation of oligonucleotide primers, since all these stages proceed sequentially and without separation of each process in time. The possibility of performing amplification under isothermal conditions largely depends on the structure and number of primers, use of special enzymes and other auxiliary proteins in the reaction mixture, and careful selection of other reaction components [3]. Notably, the principle of some variants of isothermal amplification, not without significant modifications, was inspired from various mechanisms of viral genome replication. Thus, historically, the first method of isothermal amplification was the 3SR (self-sustained sequence replication) method, which appeared owing to the studies on modeling of retroviral replication in vitro [4]. Rolling circle amplification (RCA) [5] is based on replication of viroids and some viruses with a circular genome [6–8]. Loop-mediated isothermal amplification (LAMP) has many structural homologies with the way the poxvirus genome replicates [9, 10]. In this regard, the importance of fundamental research on prokaryotic and viral replication enzymes is obvious, since the discovery of new thermostable, precise, and processive reverse transcriptases and DNA polymerases can bring LAMP technology to a completely different level of practical application, as had happened once in the field of PCR with the discovery DNA polymerase I from Thermus aquaticus, or Taq polymerase [11].
The LAMP method turned out to be the most popular method of isothermal amplification for a number of reasons: the small number of components of the reaction mixture, their availability for routine practice, and also high specificity of the reaction, which is provided by 4–6 primers [12, 13]. The simplicity of the amplification reaction allowed running LAMP under the most limited conditions on board the International Space Station [14]. Even though the LAMP technology still has disadvantages associated with currently used enzymes, the method has some advantages over PCR, which are especially valuable for clinical practice. The LAMP technology has found many applications of in-field diagnostics, when it is necessary to perform amplification outside the laboratory [15–19].
This review considers the possibilities of the LAMP method and some of its features that must be taken into account when developing new test systems. Many of the methods mentioned in the review have been tested by the authors during the development of LAMP-based assays. Corresponding theoretical and practical aspects of the method were illustrated using a specific set of primers as an example (Table 1) [20]. For the first time, the results of molecular modeling of the spatial structure of dumbbell-shaped starting structures formed during the LAMP reaction are demonstrated. The procedures for the selection of target genes and primer design are described in detail; the importance of such a stage as the prediction of unwanted secondary structures in the nucleotide sequences of the target and amplicon is noted. The main stages of optimizing the composition of the reaction mixture and amplification conditions are considered; the main characteristics of the enzymes used in the LAMP method are given; and methods for detecting a positive amplification signal and increasing the specificity of the reaction are described.
PRINCIPLE OF THE METHOD
The LAMP method was developed and patented by a group of Japanese researchers from the Eiken Chemical Co. LTD in 1998 [21]. One of the main goals of creating a new amplification method was to increase the specificity of detection of nucleotide polymorphisms in comparison with the capabilities of PCR. Currently, the patent for LAMP technology has lost its protective force, which partly explains the increased interest in this amplification method.
The reaction mechanism of LAMP with many intermediate DNA secondary structures is provided by the unusual structure of primers and resembles the Japanese art of origami [9]. For amplification by the LAMP method, it is necessary to find six annealing sites in the target gene for two pairs of primers, which, with an average length of one annealing site of 20 nt, would cover ~120 bp of the target gene. This increases the potential analytical specificity of the LAMP method by at least three times when compared to PCR based on forward and reverse primers.
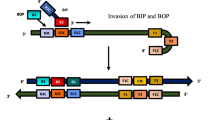
The LAMP method is based on a special structure of inner primers—forward inner primer, FIP, and backward inner primer, BIP—involved in the formation of two terminal inverted repeats in the nucleotide sequence of the amplicons. The second pair of primers—outer primers F3 and B3—is involved in amplification only at the first stages; therefore, their concentration is significantly lower than that of inner primers [10]. Due to sequential annealing of inner and outer primers, at the initial stages of the LAMP reaction the amplicon forms two types of dumbbell-shaped starting structures (Fig. 1), each having two single-stranded loops and one annealing site for the complementary inner primer. To accelerate amplification, the developers of the method suggested using the third pair of loop primers—loop forward, LF, and loop backward, LB—the annealing sites of which are also located in the loop regions of the starting structures between the F1/B1 and F2/B2 sites [22]. However, it should be taken into account that the probability of detecting annealing sites of loop primers directly depends on the length of the corresponding loops of the starting structures. For some sets of “core” primers, which is a combination of inner and outer primers [23], only one loop primer can be designed, while for others, none. It is for this reason that not every test system can be supplemented with loop primers.

Molecular models of the starting structures formed in the LAMP reaction based on the DAT primer set (Table 1). Modeling of the DNA structure was performed at a temperature of 65°C, concentration of sodium ions, 50 mM, and magnesium ions, 8 mM, using the mFold and RNAComposer software. The tertiary (left) and secondary (right) structures of amplicons are shown. The secondary structure of the amplicons shows all annealing sites for inner and loop primers, as well as their complementary regions. The length of the nucleotide sequence is 176 nt, GC-content: 55%. (a) Starting structure F, named due to the presence of an annealing site (F2c) for the FIP primer; (b) starting structure B with annealing site (B2c) for the BIP primer.
The amplification process based on the LAMP method occurs under isothermal conditions (60–65°C) and usually takes no more than 15–30 min; at a high concentration of the target nucleic acid, the reaction can reach a plateau in 5–7 min. Instead of thermal denaturation of the DNA molecule, the LAMP reaction uses the strand-displacement activity of a large fragment of the Bst DNA polymerase of Geobacillus stearothermophilus (Bst LF). Meanwhile, continuous annealing of inner and loop primers is due to the single-stranded state of the corresponding annealing sites and their constant steric accessibility for primers. At a temperature of 65°C, which is optimal for the Bst LF enzyme, the DNA double helix becomes locally denatured due to the effect of DNA “breathing” [24, 25], facilitating primer annealing at the initial stages. The strand-displacement activity of Bst LF allows the release of a previously synthesized DNA strand, which hybridizes to itself via complementary regions and forms a new DNA synthesis initiation point. The resulting starting structures, after annealing of the appropriate inner primer, can repeatedly serve as a template for the synthesis of a complementary version of themselves without the need for the original nucleic acid added to the reaction mixture. Thus, the LAMP method is a biochemical chain reaction in which the number of amplicons increases exponentially.
The rapid accumulation of DNA products in the LAMP method is provided mainly by the nucleotide sequences of inner and loop primers. When visualized in an agarose gel, a set of amplicons of different lengths forms a characteristic pattern in the form of a regular “ladder,” smoothly turning into a “smear” of high-molecular-weight amplification products (Fig. 2). During the LAMP reaction, up to 10 µg of DNA is formed per 25 µL of the reaction mixture, which is approximately two orders of magnitude higher than the total yield of PCR [26]. Therefore, when handling the contents of test tubes after the LAMP reaction, one should be aware of the possible contamination of the laboratory and reagents.

Visualization of LAMP reaction products based on the DAT primers using agarose gel electrophoresis [20]. (a) Characteristic “ladder” of amplification reaction products with typical periodicity of molecular weight (bp). M, DNA molecular mass marker (MassRuler DNA Ladder Mix, ThermoFisher Scientific, Lithuania); N, no template control; (1–3) reaction with the genomic DNA of different bacterial strains of Dickeya solani as a template; (4–9) reactions with the addition of genomic DNA of the Pectobacterium spp., found on potatoes together with D. solani, as a template; (b) Histogram of DNA band localization and molecular weights of amplification products (bp) calculated using the Vision-Capt (Vilber Lourmat, France) software.
An interesting feature of the LAMP reaction is that the amplicon does not necessarily include a nucleotide sequence flanked by primers. While in PCR the forward and reverse primers always flank the region of the gene selected for amplification, in the case of LAMP, there can be no nucleotide between the F1 and B1 sites. With this configuration of inner primers, fragments of the original target gene are retained only in the annealing sites of the loop primers (regions between F1 and F2, as well as between B1 and B2). In this regard, it should be emphasized that the LAMP method is of little use for genetic engineering and is largely adapted for diagnostics.
Since the same nucleotide sequence is repeatedly present in the composition of amplicons of different lengths [27, 28], the analysis of melting curves can be used as a characteristic of the specificity of the reaction, tracking the melting temperature (Tm) of the resulting amplification product, which, after having been measured, is reproducible for a particular set of primers. The presence of at least one restriction site out of primer sequences in the amplicon makes it possible to obtain only one band with a characteristic molecular weight in the agarose gel [9] and thus evaluate the specificity of the reaction. Any deviations in the number of peaks of melting of the LAMP reaction products, in the Tm value obtained, in the regularity of the “ladder” of amplicons in the agarose gel, or in the number of bands after the treatment of the product DNA with restriction endonucleases, indicate the presence of background nonspecific amplification in the reaction mixture, the reasons for which will be considered below.
OLIGONUCLEOTIDE PRIMERS
The determining factor for high specificity and sensitivity of detection in amplification methods is the design of oligonucleotide primers. The LAMP method based on the synchronous operation of 4–6 primers requires special attention to their sequences. If we consider LAMP as a multiplex reaction of several pairs of primers, then the effect of various thermodynamic characteristics of primers, such as the stability of hybridization of growing 3' ends and the predisposition of primers to dimerization, becomes obvious. Under conditions of constant enzymatic activity of Bst LF, dimerization of the 3' ends of the primers promotes their elongation and, consequently, the appearance of a new nucleotide sequence in the primers. Extended primers can unpredictably affect the efficiency and specificity of the reaction. Therefore, when signs of nonspecific background amplification are detected, a shift of the annealing sites of problematic primers by several nucleotides helps to reduce or eliminate complementarity in putative dimers in some cases [29]. The applicability of this approach to reduce background amplification has been experimentally shown; however, when evaluating potential dimers, one should keep in mind the existence of complementarity not only between the A·T and G·C pairs, which are usually predicted in silico, but also the noncanonical nucleotide pairs which thermodynamically contribute to primer hybridization [30].
The standard primer configuration for the LAMP reaction, consisting of core and loop primers, has been revised over time, and new types of primers have appeared.
(1) Stem primers (stem forward, StF, and stem backward, StB), whose annealing sites are located in the interval between the F1 and B1 sites [31]. Primers of this type, as in the case of loop primers, strongly depend on the length of the nucleotide sequence flanked by the F1 and B1 sites.
(2) Swarm primers F1S/B1S, which anneal to the F1c/B1c sites [32]. Note that identical annealing sites were later used by other authors for primers called FC/RC [33]. Decreasing the distance between the F2/B2 and F1/B1 annealing sites can significantly decrease nonspecific background amplification and increase analytical sensitivity [33]. The advantage of the swarm primers is that their annealing sites do not need to be searched for separately, because they coincide with the annealing sites of the inner primers.
As one can see, modification of the primer architecture and physical distance between their annealing sites are additional parameters in the optimization of the specificity or sensitivity of the method. Also, each additional pair of primers increases the multiplexity of the reaction and requires a preliminary in silico check for compatibility with other oligonucleotides to exclude possible dimers.
Evolution of the LAMP method in relation to the composition of primers, apparently, occurs not only along the path of creating new types of primers. In a recent work, the authors managed to carry out amplification based only on the inner primers without adding the outer primers, while the rest of the composition of the reaction mixture remained unchanged [34].
The selected target and amplicon, being in the single-stranded state at some stages of the reaction, should not contain undesirable secondary structures at the primer annealing sites that reduce the efficiency of amplification. The main contribution to the formation of secondary structures is made by the uneven distribution of the GC-content and, consequently, a local increase in Tm in such DNA regions. Prediction of the secondary and tertiary structures of single-stranded nucleic acids (both RNA and DNA) can be carried out upon approaching the optimal temperature and magnesium ion concentrations for Bst LF using the mFold (http://www.unafold.org) [35] and RNAComposer (http://rnacomposer.ibch.poznan.pl) web services [36]. Thus, each potential primer set and its physicochemical properties should be carefully tested using bioinformatics methods.
Difficulties in the design and compatibility of different sets of primers are perhaps the most important disadvantage of the LAMP method, which complicates the development of multiplex test systems [37–40]. Considering these features, it should be noted that today the best option for LAMP diagnostics is a test system consisting of no template control, a positive control based on plasmid DNA or armored RNA, and, finally, an optimized reaction mixture.
Many criteria for screening targets in the genome and selecting primers for LAMP have been formalized in programming languages, which has led to the creation of new highly efficient tools for solving various problems of molecular diagnostics. To simplify the design of LAMP primers, several convenient web services have been created; they allow a preliminary analysis of potential target genes, and then use the information obtained to design primers with the required parameters. Among these services are the PrimerExplorer (http://primerexplorer.jp/e) [10] by a Japanese company Eiken Chemical Co. LTD, as well as the LAMP Primer Design Tool (https://lamp.neb.com) [13] of an American company New England Biolabs (NEB). These programs make it possible to obtain candidate primer sets for the nucleotide sequence of interest, have options for sorting primers by GC-content and other physicochemical parameters (for example, by the thermodynamic stability of the 3' ends or the risk of dimerization). Despite the similarities between these web services, PrimerExplorer has some advantages. For example, it provides a number of unique possibilities for the design of primers with varying degrees of specificity for homologous nucleotide sequences. The service allows you to manually select those nucleotides that, when located at the ends of the primers, will increase their specificity for a particular taxon. If such nucleotide positions are in the middle of the primer sequence, this will lead to an increase in the tolerance of the test system, i.e., primer annealing will be more efficient despite differences in the nucleotide sequences of closely related target genes.
In the scientific literature, primer design techniques are often found, when, after selecting a target gene, homologous genes in all strains of the target pathogen are analyzed, and then multiple alignment is performed to determine conservative regions without polymorphisms [41, 42]. In this approach, however, some areas of the multiple alignments that are identified as conservative but may not actually contain species-specific nucleotides. Thus, in the process of primer design, it is necessary to include sequences of closely related orthologous genes in the multiple alignment in order to supplement the conserved primer regions with only species-specific nucleotides that are not present in the orthologous genes.
To eliminate the need to manually map multiple alignments, we developed the MorphoCatcher program [43] and online service (http://morphocatcher.ru) [44]. This service allows extracting information about the localization of taxon-specific nucleotides from multiple alignment and selecting the most polymorphic compared to orthologues region of the target gene. The performance of the service is limited only by multiple alignment algorithms, so it is useful at the stage of both screening individual orthologous genes and analysis of multiple alignment of complete viral genomes.
Recently, there has been a trend to combine primer design and screening tools with international databases of human virus genomes as part of the global GISAID initiative (https://www.gisaid.org) [45], which has led to the emergence of high-performance analytical services, such as COVID-19 CG (https://covidcg.org) [46] and Primer Monitor Tool (https://primer-monitor.neb.com) [47]. Over time, primer design algorithms for LAMP will become more user-friendly, take into account the many thermodynamic parameters of oligonucleotides, their secondary and tertiary structure, and also search for suitable targets for diagnostics based on up-to-date data from around the world.
ENZYMES FOR THE LAMP METHOD
Historically, the first and most frequently used enzyme in LAMP technology was a large fragment of DNA polymerase I from the thermophilic bacterium Geobacillus stearothermophilus (previously, the species was assigned to the Bacillus genus [48, 49], which is now reflected in the name of the enzyme). The Bst LF enzyme has 5'→3' polymerase, strand displacement, and reverse transcriptase activities, but lacks 5'→3' exonuclease activity [50, 51]. Over time, homologues of Bst LF were developed with point substitutions in the amino acid sequence, which increased the processivity, thermal stability, and reverse transcriptase activity of the enzyme at temperatures >65°C [13, 52].
The use of the technology of oligonucleotide aptamers [53] allows for reversible inhibition of the polymerase activity of Bst polymerase at room temperature, which increases the specificity of reactions based on the Bst 2.0 WarmStart enzyme (NEB, USA) [13, 23]. Thus, Bst polymerase homologues currently exist that are compatible with the hot start amplification format widely used in modern PCR applications. While Taq polymerase is reversibly inactivated by the TP7 antibody [54], which dissociates during the first stage of DNA denaturation, specific oligonucleotide aptamers are used to inhibit the Bst polymerase; they dissociate from the enzyme at temperatures >45°C [13].
Further studies have shown that DNA polymerases from other prokaryotes and viruses are also applicable to the LAMP method. Examples include a thermostable OmniAmp DNA polymerase with reverse transcriptase activity of the PyroPhage 3173 bacteriophage from the hot springs of Yellowstone National Park [55, 56], as well as a large fragment of DNA polymerase I from a soil bacterium Ureibacillus thermosphaericus [57].
When using RNA molecules as a template, as in the case of the PCR method, it is necessary to include a reverse transcription step in the amplification protocol and add one of the following enzymes to the reaction mixture:
(1) Reverse transcriptase from myeloblastosis-associated satellite virus (MAV), the origin of which is often erroneously attributed to the avian myeloblastosis virus (AMV) [58].
(2) Moloney murine leukemia virus (MMLV) reverse transcriptase [59].
(3) Hot start reverse transcriptase WarmStart RTx (NEB, USA), which have activity at 65°C [13].
As an alternative, DNA polymerase Bst 3.0 (NEB, USA) with strong revertase activity can be used for test systems with an RNA target [13].
When using these reverse transcriptases in the LAMP method, it is important to keep in mind their differences in processivity, stability, and the temperature range of activity, which does not always overlap with the temperature optimum of Bst polymerase [59]. Thus, when AMV/MAV and MMLV reverse transcriptases are combined with the LAMP method, there is a need for two temperatures for reverse transcription and amplification itself, which makes the very concept of the isothermal test somewhat contradictory since, in this case, it will require a programmable thermostat or a thermal cycler. In this regard, the use of reverse transcriptases capable of performing RNA-dependent DNA synthesis at the temperature optimum of the DNA polymerase used is justified [13, 60].
In some diagnostic tasks, when pathogen identification with a low taxonomic resolution is acceptable [47], the approach is justified, aimed at reducing the specificity of the test system to nucleotide polymorphisms in the target gene. The mismatch-tolerant amplification was first shown for PCR [61], and after some time, this approach was adapted for LAMP [62]. The technique involves the use of two polymerases in the reaction mixture: classical Bst polymerase and DNA polymerase with 3'→5' exonuclease activity. The second enzyme removes all mismatched bases from the growing ends of primers. Such a modification of the LAMP method has already found application in the development of test systems for detection of dengue virus [62] and SARS-CoV-2 coronavirus [63].
Other enzymes successfully adapted for the LAMP method include the UvrD helicase [64, 65], which can reduce the background amplification, as well as uracil-DNA glycosylase [66], which is used in PCR technology to prevent contamination by amplification products.
NEW DNA POLYMERASES
The era of the search for new wild-type enzymes to solve urgent problems of biotechnology is gradually fading into the past. Modern methods of engineering enzymology and methods for predicting the tertiary structure of proteins, which allow achieving changes in their activity with the help of single modifications of the amino acid sequence, come to the fore [67–69]. The use of such approaches helped to design thermostable homologues of Bst LF, which tolerate heating up to 90°C for 2 min, surpassing the existing commercial enzymes by this criterion (Table 2) [70–72]. The stability of the tertiary structure of Bst LF homologues can be useful for both rapid protocols of direct amplification based on thermal lysis of pathogen membranes [73] and creation of dry reaction mixtures using lyophilization or vacuum drying protocols [74, 75]. In addition, the rational design of Taq polymerase opened up a new area of application for this enzyme in isothermal amplification (Table 2). Thus, methods of directed evolution [81, 82] become essential tools for adapting known and new enzymes to a wide range of highly specialized problems of molecular diagnostics.
Interestingly, the search for strand-displacing DNA polymerases is possible not only in the genomes of the inhabitants of geothermal springs, but also among Arctic microorganisms. DNA polymerase from a marine psychrophilic bacterium Psychrobacillus sp. (PB polymerase) with an optimum activity at 25–37°C is the example. It requires only one modification (Table 2) to be compatible with the LAMP method [57]. Nevertheless, in its native form, low-temperature PB polymerase can become the basis for test systems that use the heat of the human body as a source of constant temperature. Prototypes of such test systems based on the RPA (recombinase polymerase amplification) method were developed for the diagnosis of phytopathogenic viruses [83]. The search for other DNA polymerases with a low temperature optimum of activity can lead to the development of amplification methods that do not require heating devices, which may be required when organizing mobile diagnostic stations.
Thus, an ideal DNA polymerase for LAMP amplification, apparently, should combine several properties at once: the strong strand displacement activity, high rate of DNA synthesis and processivity of the enzyme, the presence of reverse transcriptase activity, and the absence of exonuclease activity, as well as increased resistance to inhibitors of amplification.
OPTIMIZATION OF AMPLIFICATION
When developing and optimizing LAMP-based assays, an important step is the screening of primers without background amplification and false positive response in no template control. In this case, the influence of a particular factor or component of the reaction mixture must be controlled both by the amplification signal and by its specificity. When the amplification signal is generated the fastest, the contribution of background amplification may go unnoticed. In the case of intercalating dyes, it can only be detected by melting curve analysis of the final amplification product.
Optimization of LAMP amplification includes determination of the incubation temperature for the reaction (Fig. 3a) and the required concentration of magnesium ions (Fig. 3b), as well as selection of the concentrations of all primer pairs that are planned to be included in the reaction mixture (Fig. 4). The use of various amplification enhancers, commonly applied in PCR optimization, as additional components of the reaction mixture is also acceptable when working with the LAMP method. For each new component of the reaction mixture, it is necessary to demonstrate its positive effect on the amplification efficiency. For example, by adding guanidine hydrochloride to the buffer, the LAMP reaction can be significantly accelerated and the sensitivity of primers for detecting RNA or DNA can be increased [84]. An example of an additive producing a negative effect on the rate of the LAMP reaction is betaine, which lowers the melting point of G·C pairs [85] and inhibits nonspecific and specific amplification, which is confirmed by some authors and our observations (Fig. 5) [23, 86].

Effect of the temperature (a) and magnesium ion concentration (b) on the speed (left) and specificity (right) of the LAMP reaction based on the DAT primer set. RFU, relative fluorescence units. Each optimization factor was tested in three technical repeats (n = 3). Amplification curves are given in a logarithmic scale. Fluorescence signals were measured using a CFX96 Touch (Bio-Rad, USA) thermal cycler and EvaGreen (Biotium, USA) intercalating dye. The melting curves of the final amplicons only in the case of elevated temperature (66°C) and magnesium ion concentration (10 mM) allow one to notice specificity change amplification by the appearance of minor peaks or the shift of the main melting peaks. No template controls are shown in gray.

Effect of the loop primers to the reaction mixture on the speed (left) and specificity (right) of the LAMP reaction. CP, core primers (FIP, BIP, F3, and B3); LF, forward loop primer; LB, backward loop primer; LP, forward and backward loop primers. (a) Amplification curves in a logarithmic scale. Each loop primer at a concentration of 0.8 mM is capable of accelerating amplification; therefore, when two primers are added, a pronounced synergistic effect on the reaction speed is observed. (b) Melting curves of the final amplicons with identical Tm, indicating that the level of specificity is retained with an increase in the reaction speed.

Effect of betaine on the speed (left), as well as the specificity and melting point (right) of the amplification products in the LAMP reaction. (a) Amplification curves in a logarithmic scale. With an increase in the concentration of betaine, inhibition of the reaction is observed. (b) Melting curves of the final amplicons. There is a gradual decrease in Tm of the amplification products with an increase in the concentration of betaine in the reaction mixture.
When developing multicomponent reaction mixtures that require finding a balanced combination of all reagents, there is a way to significantly reduce the number of necessary experiments to find the optimal values of factors that affect the efficiency of amplification. To do this, it is necessary to design experiments using the method of orthogonal arrays, the principle of which was originally proposed by the Japanese engineer Genichi Taguchi to optimize production processes and control product quality [87, 88]. When applied to amplification methods, the Taguchi orthogonal array will contain information on the minimum number of combinations of various factors, which allows a small number of experimental tests to most widely assess the landscape of optimal values of each factor of interest by measuring the signal-to-noise ratio.
The design of experiments based on the classical factorial array in the presence of five optimization factors (for example, temperature and four components of the reaction mixture) in three different variable values prescribes to experimentally test 243 (35) combinations. The Taguchi method allows for a decrease of the number of necessary experiments to 27. The mathematical model created on the basis of the dispersion analysis of experimental data makes it possible to determine the optimal values of the factors, as well as to calculate the percentage contribution of each factor to the amplification efficiency [89]. Despite the obvious advantages of this approach in the development of test systems, the Taguchi method is used undeservedly rarely.
DETECTION OF AMPLIFICATION
The LAMP method proved to be compatible with detection methods previously used in PCR, such as agarose gel electrophoresis and real-time amplification detection with intercalating dyes [9]. In order to identify suitable intercalating dyes (for example, some dyes of the SYTO family, EvaGreen, etc.), a series of comparative studies of their inhibitory effect on the LAMP reaction was carried out [90, 91]. Later, for LAMP detection, a technique was proposed based on labeling of inner and loop primers for subsequent visualization of the amplification product using lateral flow immunochromatographic assays [92, 93]. Due to the peculiarities of the reaction, fundamentally new methods of visual detection were developed for LAMP, which are currently not available in PCR technology. Let us consider these detection methods in more detail.
The first qualitative sign of a positive LAMP reaction was found to be the formation of a white insoluble precipitate of magnesium pyrophosphate at the bottom of the test tubes. It was proposed to evaluate the dynamics of the increase in the turbidity of the reaction mixture using portable thermostats specially designed for this purpose [26]. Thus, an additional sign of the positive LAMP reaction was a decrease in the concentration of free magnesium ions in the reaction mixture.
Visualization of the LAMP reaction using magnesium is based on the divalent metal ion indicators, which, when the concentration of free ions decreases, can change the color of the reaction mixture in visible or UV light [94, 95]. Historically, the first such metal indicator was calcein [10], whose action requires the addition of manganese ions to the reaction mixture. A little later, the hydroxynaphthol blue dye (Fig. 6d), which is widely used for mobile diagnostics and does not require additional modifications of the reaction mixture composition, was introduced [96]. Recently, the eriochrome black T indicator has been gaining popularity, especially in the LAMP detection in microfluidic chips [74, 97]. Another metal indicator, known as acid chrome blue K, was also successfully tested for LAMP detection [98].

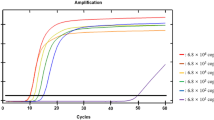
Analytical sensitivity of the DAT primer set in the concentration range of 4 × 100–4 × 105 copies of D. solani genomic DNA during the LAMP reaction. (a) Amplification curves in a logarithmic scale; (b) melting curves of the final amplicons; and (c) standard curve with visualization of the linear dynamic range and the limit of detection. The amplification efficiency was 99.4%, R2 = 0.948. (d) Visualization of LAMP by hydroxynaphthol blue. N, no template control; (1–6) reactions with the addition of genomic DNA in descending order of concentration. At the bottom of test tubes with positive reactions (1–5), a precipitate of magnesium pyrophosphate is visible. The limit of detection (40 cp/rxn) based on hydroxynaphthol blue is the same as that obtained in the real-time detection format.
An alternative method for detecting LAMP is based on the use of various pH-sensitive dyes that change color when the reaction mixture is acidified during DNA synthesis. This group of indicators includes phenol red, cresol red, neutral red, and metacresol purple [99]. Recently, the use of xylenol orange [100] and some fluorescein derivatives [101] for LAMP detection was shown.
When using the dye indicators described above, we are dealing with indirect detection of amplification, since the by-products of the LAMP reaction, magnesium pyrophosphate and hydrogen ions, play an important role in their operation. Therefore, the presence of various interfering substances (for example, divalent metal ions) in the sample or an inappropriate sample pH may cause the LAMP reaction mixture to change color prior to amplification. Thus, even atmospheric carbon dioxide can have a negative effect on the shelf life of reagents based on phenol red, acidifying a weakly buffered reaction mixture [102].
Berberine [103], crystal violet [104], malachite green [105], and methyl green [106] are indicators that change their color in response to an increase in the concentration of amplified DNA. Summing up, we note that the use of any type of visual detection in clinical diagnostics requires the most objective estimates of color changes based on the spectrophotometric analysis [84, 102].
An urgent task is still an improvement of ways for direct detection of LAMP using a special structure of primers labeled with a fluorophore and a fluorescence quencher [12]. The absence of 5'→3' exonuclease activity in Bst LF makes it difficult to use TaqMan probes, as well as molecular beacons [107, 108] in the LAMP method for real-time detection of amplification. The TaqMan technology works in LAMP with some background amplification in no template controls [109] while molecular beacons are characterized by extremely low specificity [110]. Therefore, labeling inner primers with a fluorophore seems to be the optimal solution to the problem of direct detection [37].
LAMP IN QUANTITATIVE ANALYSIS
The advantage of quantitative analysis of nucleic acids based on PCR is that the doubling of the number of amplicons under optimized reaction conditions, under which the amplification efficiency is ~100%, is strictly related to the thermal cycling steps and occurs in each new cycle [111]. In the LAMP reaction, the kinetic parameters of primer annealing and elongation can vary greatly, since they are determined by the thermodynamic properties of the primers and the target gene region being amplified. Thus, the high efficiency of the LAMP reaction contributes to a decrease in the resolution of the test system on samples with slight differences in the concentration of the target nucleic acid [112]. Paradoxically, the LAMP method can be simultaneously characterized as a very sensitive method with a low detection limit, on the one hand, and a method with poor resolution with respect to estimation of the number of copies of the target in samples with less than a 10-fold difference in the starting concentration of nucleic acid. This is largely due to the fact that it takes only a few minutes to pass through the entire linear dynamic range of the LAMP reaction (Fig. 6c) [112]. While the values of the PCR threshold cycles can be recalculated with a sufficiently high accuracy into the number of copies of the target gene, in the case of the threshold time values in LAMP, we can reliably distinguish only a 10-fold difference in the concentrations of the target nucleic acid. In this regard, the applicability of the LAMP method for real-time quantitative diagnostics seems to be rather controversial, despite the proposed mathematical models of amplification kinetics [27, 113]. Does this mean that the LAMP method is absolutely inapplicable for the quantitative analysis?
To understand the answer to this question, one should consider the LAMP technology in the light of the digital PCR, the most advanced method for the quantitative analysis of nucleic acids [114, 115]. The essence of digital PCR is that the standard reaction mixture is divided into thousands of individual microreactions, each of which may contain either no or single copies of target genes. In this case, target detection occurs at the limit of detection since the multitude of individual microreactions eliminates possible differences in the concentrations of the target gene in each of them. Therefore, this makes it possible to use any sensitive amplification method for nucleic acid detection, among which the LAMP method should be considered [116].
Thus, the LAMP method can compete with PCR in digital amplification format. First, the simplest resistive heating element is sufficient for the LAMP reaction, so the cost of developing an accurate and high-speed thermal cycler can be safely deducted from the cost of a device for a digital LAMP. Secondly, the high yield of the LAMP reaction in combination with bright intercalating dyes can provide a higher fluorescence intensity of individual microreactions with a positive signal, which will allow them to be more accurately distinguished from microreactions with a negative signal. Perhaps, the digital LAMP will not have significant advantages only at the stages of development and production of chips necessary for generating droplets of the reaction mixture in oil [117] or inside individual microreactors [74].
CONCLUSIONS
The flexibility of isothermal amplification methods, with the ability to fine-tune each reaction component, is one of the driving characteristics that has contributed to the emergence of a huge number of modifications of the LAMP method. Today, many of them claim to be the “gold standard” for mobile diagnostics [118, 119]. The unpretentiousness of isothermal amplification to the temperature source and compatibility with the dry format of the reaction mixture opens up broad prospects for the LAMP method. There are many different portable platforms to perform the LAMP reaction that were produced thanks to the efforts of Russian scientific groups as well. They include:
(1) Mobile thermostats for 0.2-mL tubes with real-time detection of the amplification signal [13, 65, 120, 121].
(2) Microfluidic lab-on-a-chip platforms with compartments for nucleic acid isolation, LAMP, and amplicon detection using a lateral flow test [122].
(3) Disposable chips with many microreaction chambers and special cartridges for the LAMP reaction mixture, which allow analyzing amplification results using a smartphone [74, 97, 123].
At present, the detection of single nucleotide polymorphisms has not become the main advantage of LAMP, as planned by the developers of the method [21]. Nevertheless, an original LAMP-based strategy for allele-specific detection has been proposed employing two oligonucleotide probes, each of which represents half of the starting structure [124]. Annealing of such probes on a single nucleotide polymorphism leads to probe ligation and the formation of a complete starting structure that triggers the LAMP reaction only if the desired nucleotide substitution is present in the target gene. Perhaps the development of this particular amplification method will help in the adaptation of the LAMP method to research in the field of genetic diseases.
Features of the starting structures and the architecture of LAMP primers served as an important starting point in the development of other methods of isothermal amplification [33, 125]. Some results seem very promising for implementation in the LAMP method in order to increase its sensitivity to nucleotide polymorphisms. In particular, the DNA-binding protein MutS from T. aquaticus is of particular interest, which became the basis for the detection of nucleotide polymorphisms in one of the asymmetric methods of isothermal amplification [126]. Improvement of primer design algorithms and rational inclusion of modified nucleotides, such as LNA and PNA, in their sequence [127, 128] can increase the storage stability of primers and lead to the emergence of more specific allele-specific LAMP methods.
The COVID-19 pandemic served as an important impetus for the development of the LAMP method, inspiring researchers to improve express sample preparation techniques [129, 130], develop direct amplification assays that do not require isolation and purification of nucleic acids [131, 132], optimize alternative DNA polymerases and reverse transcriptases [60], and create new detection methods based on various CRISPR-Cas nucleases [133–136]. It was in the recent years that large-scale testing of LAMP technology in clinical practice began [19, 102, 137].
All these studies can contribute to the gradual implementation of the LAMP method into the healthcare system as an inexpensive, specific, and rapid way to conduct screening of large populations [138]. Many difficulties are on its way to the field of molecular diagnostics, arising from the widespread implementation of PCR in diagnostic laboratories and the dominant position of PCR in legal documents governing this area in medicine and agriculture. It can be expected that it is the economic factor that will play an important role in the intensity of measures to implement LAMP technology in many countries of the world.
Thus, the LAMP method has become in many ways a revolutionary solution in the field of mobile molecular diagnostics, creating a whole galaxy of new methods for detecting nucleic acid amplification. In the future, LAMP technology has every chance of becoming a full-fledged alternative to PCR in the field of molecular diagnostics of pathogens, and new modifications of LAMP will certainly help this interesting amplification method find its unique scope of application.
REFERENCES
Esbin, M.N., Whitney, O.N., Chong, S., Maurer, A., Darzacq, X., and Tjian, R., RNA, 2020, vol. 26, pp. 771–783. https://doi.org/10.1261/rna.076232.120
Chaouch, M., Rev. Med. Virol., 2021, vol. 31, pp. e2215. https://doi.org/10.1002/rmv.2215
Islam, M.M. and Koirla, D., Anal. Chim. Acta, 2021, p. 339338. https://doi.org/10.1016/j.aca.2021.339338
Guatelli, J.C., Whitfield, K.M., Kwoh, D.Y., Barringer, K.J., Richman, D.D., and Gingeras, T.R., Proc. Natl. Acad. Sci. U.S.A., 1990, vol. 87, pp. 1874–1878. https://doi.org/10.1073/pnas.87.5.1874
Mohsen, M.G. and Kool, E.T., Acc. Chem. Res., 2016, vol. 49, pp. 2540–2550. https://doi.org/10.1021/acs.accounts.6b00417
Kusumoto-Matsuo, R., Kanda, T., and Kukimoto, I., Genes Cells, 2011, vol. 16, pp. 23–33. https://doi.org/10.1111/j.1365-2443.2010.01458.x
Flores, R., Serra, P., Minoia, S., Di Serio, F., and Navarro, B., Front. Microbiol., 2012, vol. 3, p. 217. https://doi.org/10.3389/fmicb.2012.00217
Wawrzyniak, P., Płucienniczak, G., and Bartosik, D., Front. Microbiol., 2017, vol. 8, p. 2353. https://doi.org/10.3389/fmicb.2017.02353
Notomi, T., Okayama, H., Masubuchi, H., Yonekawa, T., Watanabe, K., Amino, N., and Hase, T., Nucleic Acid Res., 2000, vol. 28, p. e63. https://doi.org/10.1093/nar/28.12.e63
Tomita, N., Mori, Y., Kanda, H., and Notomi, T., Nat. Protoc., 2008, vol. 3, pp. 877–882. https://doi.org/10.1038/nprot.2008.57
Green, M.R. and Sambrook, J., Cold Spring Harb. Protoc., 2019, vol. 2019, pp. 436–456. https://doi.org/10.1101/pdb.top095109
Becherer, L., Borst, N., Bakheit, M., Frischmann, S., Zengerle, R., and von Stetten, F., Anal. Methods, 2020, vol. 12, pp. 717–746. https://doi.org/10.1039/C9AY02246E
Moore, K.J.M., Cahill, J., Aidelberg, G., Aronoff, R., Bektaş, A., Bezdan, D., Butler, D.J., Chittur, S.V., Codyre, M., Federici, F., Tanner, N.A., Tighe, S.W., True, R., Ware, S.B., Wyllie, A.L., Afshin, E.E., Bendesky, A., Chang, C.B., Dela, Rosa, R., 2nd, Elhaik, E., et al., J. Biomol. Tech., 2021, vol. 32, pp. 228–275. https://doi.org/10.7171/jbt.21-3203-017
Rubinfien, J., Atabay, K.D., Nichols, N.M., Tanner, N.A., Pezza, J.A., Gray, M.M., Wagner, B.M., Poppin, J.N., Aken, J.T., Gleason, E.J., Foley, K.D., Copeland, D.S., Kraves, S., Alvarez Saavedra, E., FASEB Bioadv., 2020, vol. 2, pp. 160–165. https://doi.org/10.1096/fba.2019-00088
Tomlinson, J., Methods Mol. Biol., 2013, vol. 938, pp. 291–300. https://doi.org/10.1007/978-1-62703-089-2_25
Suea-Ngam, A., Bezinge, L., Mateescu, B., Howes, P.D., DeMello, A.J., and Richards, D.A., ACS Sens., 2020, vol. 5, pp. 2701–2723. https://doi.org/10.1021/acssensors.0c01488
Yadav, S., Sharma, N.N., and Akhtar, J., Analyst, 2021, vol. 146, pp. 3422–3439. https://doi.org/10.1039/d1an00214g
Liu, W., Yue, F., and Lee, L.P., Acc. Chem. Res., 2021, vol. 54, pp. 4107–4119. https://doi.org/10.1021/acs.accounts.1c00385
Mozsary, C., McCloskey, D., Babler, K.M., Boza, J., Butler, D., Currall, B., Williams, S., Wiley, A., Afshin, E.E., Grills, G.S., Sharkey, M.E., Premsrirut, P., Solo-Gabriele, H., Cardentey, Y., Erickson, D., and Mason, C.E., J. Biomol. Tech., 2021, vol. 32, pp. 221–227. https://doi.org/10.7171/jbt.21-3203-019
Miroshnikov, K.A. and Shirshikov, F.V., Patent RU 2642313 C1, 2018.
Notomi, T. and Hase, T., Patent US 6410278, 2002.
Nagamine, K., Hase, T., and Notomi, T., Mol. Cell. Probes, 2002, vol. 16, pp. 223–229. https://doi.org/10.1006/mcpr.2002.0415
Tanner, N.A. and Evans, T.C., Jr., Curr. Protoc. Mol. Biol., 2014, vol. 105, p. 15.14.1–15.14.14. https://doi.org/10.1002/0471142727.mb1514s105
von Hippel, P.H., Johnson, N.P., and Marcus, A.H., Biopolymers, 2013, vol. 99, pp. 923–954. https://doi.org/10.1002/bip.22347
Beyerle, E.R., Dinpajooh, M., Ji, H., von Hippel, P.H., Marcus, A.H., and Guenza, M.G., Nucleic Acids Res., 2021, vol. 49, pp. 1872–1885. https://doi.org/10.1093/nar/gkab015
Mori, Y., Nagamine, K., Tomita, N., and Notomi, T., Biochem. Biophys. Res. Commun., 2001, vol. 289, pp. 150–154. https://doi.org/10.1006/bbrc.2001.5921
Schneider, L., Blakely, H., and Tripathi, A., Electrophores., 2019, vol. 40, pp. 2706–2717. https://doi.org/10.1002/elps.201900167
Kaur, N., Thota, N., and Toley, B.J., Comput. Struct. Biotechnol. J., 2020, vol. 18, pp. 2336–2346. https://doi.org/10.1016/j.csbj.2020.08.020
Meagher, R.J., Priye, A., Light, Y.K., Huang, C., and Wang, E., Analyst, 2018, vol. 143, pp. 1924–1933. https://doi.org/10.1039/c7an01897e
SantaLucia, J., Jr. and Hicks, D., Annu. Rev. Biophys. Biomol. Struct., 2004, vol. 33, pp. 415–440. https://doi.org/10.1146/annurev.biophys.32.110601.141800
Gandelman, O., Jackson, R., Kiddle, G., and Tisi, L., Int. J. Mol. Sci., 2011, vol. 12, pp. 9108–9124. https://doi.org/10.3390/ijms12129108
Martineau, R.L., Murray, S.A., Ci, S., Gao, W., Chao, S.-H., and Meldrum, D.R., Anal. Chem., 2017, vol. 89, pp. 625–632. https://doi.org/10.1021/acs.analchem.6b02578
Ding, X., Xu, Z., Yin, K., Sfeir, M., and Liu, C., Anal. Chem., 2019, vol. 91, pp. 12852–12858. https://doi.org/10.1021/acs.analchem.9b02582
Mao, R., Wang, T., Zhao, Y., Wu, X., Zhang, S., and Cai, T., Talanta, 2022, vol. 240, p. 123217. https://doi.org/10.1016/j.talanta.2022.123217
Zuker, M., Nucleic Acid Res., 2003, vol. 31, pp. 3406–3415. https://doi.org/10.1093/nar/gkg595
Popenda, M., Szachniuk, M., Antczak, M., Purzycka, K.J., Lukasiak, P., Bartol, N., Blazewicz, J., and Adamiak, R.W., Nucleic Acid Res., 2012, vol. 40, p. e112. https://doi.org/10.1093/nar/gks339
Tanner, N.A., Zhang, Y., and Evans, T.C., BioTechniques, 2012, vol. 53, pp. 81–89. https://doi.org/10.2144/0000113902
Ball, C.S., Light, Y.K., Koh, C.-Y., Wheeler, S.S., Coffey, L.L., and Meagher, R.J., Anal. Chem., 2016, vol. 88, pp. 3562–3568. https://doi.org/10.1021/acs.analchem.5b04054
Mayboroda, O., Katakis, I., and O’Sullivan, C.K., Anal. Biochem., 2018, vol. 545, pp. 20–30. https://doi.org/10.1016/j.ab.2018.01.005
Zhang, Y. and Tanner, N.A., PLoS One, 2022, vol. 17. https://doi.org/10.1371/journal.pone.0254324
Yan, W., Methods Mol. Biol., 2015, vol. 1275, pp. 91–101. https://doi.org/10.1007/978-1-4939-2365-6_6
Skuza, L., Methods Mol. Biol., 2022, vol. 2392, pp. 83–91. https://doi.org/10.1007/978-1-0716-1799-1_6
Shirshikov, F.V., Pekov, Yu.A., Karpets, P.Yu., and Miroshnikov, K.A., Svidetel’stvo o gosudarstvennoi registratsii programmy dlya EVM RU 2018616252 (Certificate of registration of a computer software RU 2018616252), 2018.
Shirshikov, F.V., Pekov, Y.A., and Miroshnikov, K.A., PeerJ, 2019, vol. 7, p. e6801. https://doi.org/10.7717/peerj.6801
Elbe, S. and Buckland-Merrett, G., Glob. Chall., 2017, vol. 1, pp. 33–46. https://doi.org/10.1002/gch2.1018
Chen, A.T., Altschuler, K., Zhan, S.H., Chan, Y.A., and Deverman, B.E., eLife, 2021, vol. 10, p. e63409. https://doi.org/10.7554/elife.63409
Tamanaha, E., Zhang, Y., and Tanner, N.A., PLoS One, 2022, vol. 17, p. e0259610. https://doi.org/10.1371/journal.pone.0259610
Nazina, T.N., Tourova, T.P., Poltaraus, A.B., Novikova, E.V., Grigoryan, A.A., Ivanova, A.E., Lysenko, A.M., Petrunyaka, V.V., Osipov, G.A., Belyaev, S.S., and Ivanov, M.V., Int. J. Syst. Evol. Microbiol., 2001, vol. 51, pp. 433–446. https://doi.org/10.1099/00207713-51-2-433
Coorevits, A., Dinsdale, A.E., Halket, G., Lebbe, L., De Vos, P., Van Landschoot, A., and Logan, N.A., Int. J. Syst. Evol. Microbiol., 2012, vol. 62, pp. 1470–1485. https://doi.org/10.1099/ijs.0.030346-0
Oscorbin, I.P., Boyarskikh, U.A., and Filipenko, M.L., Mol. Biotechnol., 2015, vol. 57, pp. 947–959. https://doi.org/10.1007/s12033-015-9886-x
Jackson, L.N., Chim, N., Shi, C., and Chaput, J.C., Nucleic Acids Res., 2019, vol. 47, pp. 6973–6983. https://doi.org/10.1093/nar/gkz513
Oscorbin, I.P., Belousova, E.A., Boyarskikh, U.A., Zakabunin, A.I., Khrapov, E.A., and Filipenko, M.L., Nucleic Acids Res., 2017, vol. 45, pp. 9595–9610.
Xiao, X., Li, H., Zhao, L., Zhang, Y., and Liu, Z., Biomed. Pharmacother., 2021, vol. 143, p. 112232. https://doi.org/10.1016/j.biopha.2021.112232
Murali, R., Sharkey, D.J., Daiss, J.L., and Murthy, H.M., Proc. Natl. Acad. Sci. U.S.A., 1998, vol. 95, pp. 12562–12567. https://doi.org/10.1073/pnas.95.21.12562
Moser, M.J., DiFrancesco, R.A., Gowda, K., Klingele, A.J., Sugar, D.R., Stocki, S., Mead, D.A., and Schoenfeld, T.W., PLoS One, 2012, vol. 7, p. e38371. https://doi.org/10.1371/journal.pone.0038371
Chander, Y., Koelbl, J., Puckett, J., Moser, M.J., Klingele, A.J., Liles, M.R., Carrias, A., Mead, D.A., and Schoenfeld, T.W., Front. Microbiol., 2014, vol. 5, p. 395. https://doi.org/10.3389/fmicb.2014.00395
Piotrowski, Y., Gurung, M.K., and Larsen, A.N., BMC Mol. Cell Biol., 2019, vol. 20, p. 31. https://doi.org/10.1186/s12860-019-0216-1
Perbal, B., Retrovirology, 2008, vol. 5, p. 49. https://doi.org/10.1186/1742-4690-5-49
Rittié, L. and Perbal, B., J. Cell Commun. Signal., 2008, vol. 2, pp. 25–45. https://doi.org/10.1007/s12079-008-0026-2
Alekseenko, A., Barrett, D., Pareja-Sanchez, Y., Howard, R.J., Strandback, E., Ampah-Korsah, H., Rovšnik, U., Zuniga-Veliz, S., Klenov, A., Malloo, J., Ye, S., Liu, X., Reinius, B., Elsӓsser, S.J., Nyman, T., Sandh, G., Yin, X., and Pelechano, V., Sci. Rep., vol. 11, p. 1820. https://doi.org/10.1038/s41598-020-80352-8
Li, Y., Wan, Z., Hu, Y., Zhou, Y., Chen, Q., and Zhang, C., BioTechniques, 2019, vol. 66, pp. 225–230. https://doi.org/10.2144/btn-2018-0184
Zhou, Y., Wan, Z., Yang, S., Li, Y., Li, M., Wang, B., Hu, Y., Xia, X., Jin, X., Yu, N., and Zhang, C., Front. Microbiol., 2019, vol. 10, p. 1056. https://doi.org/10.3389/fmicb.2019.01056
Lu, R., Wu, X., Wan, Z., Li, Y., Zuo, L., Qin, J., Jin, X., and Zhang, C., Virol. Sin., 2020, vol. 35, pp. 344–347. https://doi.org/10.1007/s12250-020-00218-1
Bokelmann, L., Nickel, O., Maricic, T., Paabo, S., Meyer, M., Borte, S., and Riesenberg, S., Nat. Commun., 2021, vol. 12, p. 1467. https://doi.org/10.1038/s41467-021-21627-0
Natoli, M.E., Kundrod, K.A., Chang, M.M., Smith, C.A., Paul, S., Coole, J.B., Butlin, N.G., Tanner, N.A., Baker, E., Schmeler, K.M., and Richards-Kortum, R., J. Biomol. Tech., 2021, vol. 32, pp. 180–185. https://doi.org/10.7171/jbt.21-3203-013
Hsieh, K., Mage, P.L., Csordas, A.T., Eisenstein, M., and Tom Soh, H., Chem. Commun., 2014, vol. 50, pp. 3747–3749. https://doi.org/10.1039/c4cc00540f
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Zidek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S.A.A., Ballard, A.J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., Petersen, S., Reiman, D., Clancy, E., Zielinski, M., Steinegger, M., Pacholska, M., Berghammer, T., Bodenstein, S., Silver, D., Vinyals, O., Senior, A.W., Kavukcuoglu, K., Kohli, P., and Hassabis, D., Nature, 2021, vol. 596, pp. 583–589. https://doi.org/10.1038/s41586-021-03819-2
Coulther, T.A., Stern, H.R., and Beuning, P.J., Trends Biotechnol., 2019, vol. 37, pp. 1091–1103. https://doi.org/10.1016/j.tibtech.2019.03.011
Nikoomanzar, A., Chim, N., Yik, E.J., and Chaput, J.C., Q. Rev. Biophys., 2020, vol. 53, p. e8. https://doi.org/10.1017/s0033583520000050
Milligan, J.N., Shroff, R., Garry, D.J., and Ellington, A.D., Biochemistry, 2018, vol. 57, pp. 4607–4619. https://doi.org/10.1021/acs.biochem.8b00200
Paik, I., Ngo, P.H.T., Shroff, R., Diaz, D.J., Maranhao, A.C., Walker, D.J.F., Bhadra, S., and Ellington, A.D., Biochemistry, 2021. https://doi.org/10.1021/acs.biochem.1c00451
Paik, I., Bhadra, S., and Ellington, A.D., ACS Synth. Biol., 2022, vol. 11, pp. 1488–1496. https://doi.org/10.1021/acssynbio.1c00559
Hu, L.X., Yang, Z.H., Zhang, D., Zhao, D.M., and Zhu, J.H., Lett. Appl. Microbiol., 2016, vol. 63, pp. 289–296. https://doi.org/10.1111/lam.12618
Rodriguez-Manzano, J., Karymov, M.A., Begolo, S., Selck, D.A., Zhukov, D.V., Jue, E., and Ismagilov, R.F., ACS Nano, vol. 10, no. 2016, pp. 3102–3113. https://doi.org/10.1021/acsnano.5b07338
García-Bernalt Diego, J., Fernández-Soto, P., Crego-Vicente, B., Alonso-Castrillejo, S., Febrer-Sendra, B., Gomez-Sanchez, A., Vicente, B., López-Abán, J., and Muro, A., Sci. Rep., vol. 9, p. 14744. https://doi.org/10.1038/s41598-019-51342-2
Ignatov, K. and Kramarov, V., Patent US 9896671 B2, 2018.
Ignatov, K.B., Barsova, E.V., Fradkov, A.F., Blagodatskikh, K.A., Kramarova, T.V., and Kramarov, V.M., BioTechniques, 2014, vol. 57, pp. 81–87. https://doi.org/10.2144/000114198
Varlamov, D.A., Blagodatskikh, K.A., Smirnova, E.V., Kramarov, V.M., and Ignatov, K.B., Front. Bioeng. Biotechnol., 2020, vol. 8, p. 604793. https://doi.org/10.3389/fbioe.2020.604793
Barnes, W.M., Zhang, Z., and Kermekchiev, M.B., Front. Bioeng. Biotechnol., 2020, vol. 8, p. 553474. https://doi.org/10.3389/fbioe.2020.553474
Arezi, B., McKinney, N., Hansen, C., Cayouette, M., Fox, J., Chen, K., Lapira, J., Hamilton, S., and Hogrefe, H., Front. Microbiol., 2014, vol. 5, p. 408. https://doi.org/10.3389/fmicb.2014.00408
Ouaray, Z., Singh, I., Georgiadis, M.M., and Richards, N.G.J., Protein Sci., 2020, vol. 29, pp. 455–468. https://doi.org/10.1002/pro.3762
Ouaray, Z., Benner, S.A., Georgiadis, M.M., and Richards, N.G.J., J. Biol. Chem., 2020, vol. 295, pp. 17046–17059. https://doi.org/10.1074/jbc.rev120.013745
Verosloff, M., Chappell, J., Perry, K.L., Thompson, J.R., and Lucks, J.B., ACS Synth. Biol., 2019, vol. 8, pp. 902–905. https://doi.org/10.1021/acssynbio.8b00526
Zhang, Y., Ren, G., Buss, J., Barry, A.J., Patton, G.C., and Tanner, N.A., BioTechniques, 2020, vol. 69, pp. 178–185. https://doi.org/10.2144/btn-2020-0078
Vasudevamurthy, M.K., Lever, M., George, P.M., and Morison, K.R., Biopolymers, 2009, vol. 91, pp. 85–94.
Ma, C., Wang, Y., Zhang, P., and Shi, C., Anal. Biochem., 2017, vol. 530, pp. 1–4. https://doi.org/10.1016/j.ab.2017.04.017
Cobb, B.D. and Clarkson, J.M., Nucleic Acids Res., 1994, vol. 22, pp. 3801–3805. https://doi.org/10.1093/nar/22.18.3801
Thanakiatkrai, P. and Welch, L., Int. J. Legal Med., 2012, vol. 126, pp. 161–165. https://doi.org/10.1007/s00414-011-0558-5
Morero, M., Ramirez, M.R., and Oyhenart, J., Vet. Parasitol., 2021, vol. 295, p. 109462. https://doi.org/10.1016/j.vetpar.2021.109462
Oscorbin, I.P., Belousova, E.A., Zakabunin, A.I., Boyarskikh, U.A., and Filipenko, M.L., BioTechniques, 2016, vol. 61, pp. 20–25. https://doi.org/10.2144/000114432
Quyen, T.L., Ngo, T.A., Bang, D.D., Madsen, M., and Wolff, A., Front. Microbiol., 2019, vol. 10, p. 2234. https://doi.org/10.3389/fmicb.2019.02234
Phillips, E.A., Moehling, T.J., Bhadra, S., Ellington, A.D., and Linnes, J.C., Anal. Chem., 2018, vol. 90, pp. 6580–6586. https://doi.org/10.1021/acs.analchem.8b00269
Li, J., Macdonald, J., and von Stetten, F., Analyst, 2019, vol. 144, pp. 31–67. https://doi.org/10.1039/c8an01621f
Scott, A.T., Layne, T.R., O’Connell, K.C., Tanner, N.A., and Landers, J.P., Anal. Chem., 2020, vol. 92, pp. 13343–13353. https://doi.org/10.1021/acs.analchem.0c02666
Varona, M. and Anderson, J.L., Anal. Chem., 2019, vol. 91, pp. 6991–6995. https://doi.org/10.1021/acs.analchem.9b01762
Goto, M., Honda, E., Ogura, A., Nomoto, A., and Hanaki, K.-I., BioTechniques, 2009, vol. 46, pp. 167–172. https://doi.org/10.2144/000113072
Nguyen, H.Q., Nguyen, V.D., Van Nguyen, H., and Seo, T.S., Sci. Rep., vol. 10, p. 15123. https://doi.org/10.1038/s41598-020-72095-3
Wang, X., Fu, Z., Chen, X., Peng, C., Xu, X., Wei, W., Li, F., and Xu, J., Anal. Bioanal. Chem., 2017, vol. 409, pp. 881–889. https://doi.org/10.1007/s00216-016-0084-x
Tanner, N.A., Zhang, Y., and Evans, T.C., BioTechniques, 2015, vol. 58, pp. 59–68. https://doi.org/10.2144/000114253
Jaroenram, W., Cecere, P., and Pompa, P.P., J. Microbiol. Methods, 2019, vol. 156, pp. 9–14. https://doi.org/10.1016/j.mimet.2018.11.020
Brown, T.A., Schaefer, K.S., Tsang, A., Yi, H.A., Grimm, J.B., Lemire, A.L., Jradi, F.M., Kim, C., McGowan, K., Ritola, K., Armstrong, D.T., Mostafa, H.H., Korff, W., Vale, R.D., and Lavis, L.D., J. Biomol. Tech., vol. 32, no. 3, 2021, pp. 121–133. https://doi.org/10.7171/jbt.21-3203-007
Dao Thi, V.L., Herbst, K., Boerner, K., Meurer, M., Kremer, L.P., Kirrmaier, D., Freistaedter, A., Papagiannidis, D., Galmozzi, C., Stanifer, M.L., Boulant, S., Klein, S., Chlanda, P., Khalid, D., Barreto, Miranda, I., Schnitzler, P., Krausslich, H.G., Knop, M., and Anders, S., Sci. Transl. Med., 2020, vol. 12. eabc7075. https://doi.org/10.1126/scitranslmed.abc7075
Fischbach, J., Xander, N.C., Frohme, M., and Glökler, J.F., BioTechniques, 2015, vol. 58, pp. 189–194. https://doi.org/10.2144/000114275
Miyamoto, S., Sano, S., Takahashi, K., and Jikihara, T., Anal. Biochem., 2015, vol. 473, pp. 28–33. https://doi.org/10.1016/j.ab.2014.12.016
Lucchi, N.W., Ljolje, D., Silva-Flannery, L., and Udhayakumar, V., PLoS One, 2016, vol. 11, p. e0151437. https://doi.org/10.1371/journal.pone.0151437
Thapa, J., Maharjan, B., Malla, M., Fukushima, Y., Poudel, A., Pandey, B.D., Hyashida, K., Gordon, S.V., Nakajima, C., and Suzuki, Y., Tuberculosis, 2019, vol. 117, pp. 1–6. https://doi.org/10.1016/j.tube.2019.05.004
Liu, W., Huang, S., Liu, N., Dong, D., Yang, Z., Tang, Y., Ma, W., He, X., Ao, D., Xu, Y., Zou, D., and Huang, L., Sci. Rep., vol. 7, p. 40125. https://doi.org/10.1038/srep40125
Sherrill-Mix, S., Hwang, Y., Roche, A.M., Glascock, A., Weiss, S.R., Li, Y., Haddad, L., Deraska, P., Monahan, C., Kromer, A., Graham-Wooten, J., Taylor, L.J., Abella, B.S., Ganguly, A., Collman, R.G., Van Duyne, G.D., and Bushman, F.D., Genome Biol., 2021, vol. 22, p. 169. https://doi.org/10.1186/s13059-021-02387-y
Liang, R., Liang, L., Ren, X., Jia, Y., Han, K., Zhao, J., Song, C., and Cui, S., Arch. Virol., 2021, vol. 166, pp. 1599–1605. https://doi.org/10.1007/s00705-021-04963-w
Hardinge, P. and Murray, J.A.H., BMC Biotechnol., 2019, vol. 19, p. 55. https://doi.org/10.1186/s12896-019-0549-z
Bustin, S.A., Benes, V., Garson, J.A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M.W., Shipley, G.L., Vandesompele, J., and Wittwer, C.T., Clin. Chem., 2009, vol. 55, pp. 611–622. https://doi.org/10.1373/clinchem.2008.112797
Schoepp, N.G., Schlappi, T.S., Curtis, M.S., Butkovich, S.S., Miller, S., Humphries, R.M., and Ismagilov, R.F., Sci. Transl. Med., 2017, vol. 9, p. eaal3693. https://doi.org/10.1126/scitranslmed.aal3693
Subramanian, S. and Gomez, R.D., PLoS One, 2014, vol. 9, p. e100596. https://doi.org/10.1371/journal.pone.0100596
Huggett, J.F., Foy, C.A., Benes, V., Emslie, K., Garson, J.A., Haynes, R., Hellemans, J., Kubista, M., Mueller, R.D., Nolan, T., Pfaffl, M.W., Shipley, G.L., Vandesompele, J., Wittwer, C.T., and Bustin, S.A., Clin. Chem., 2013, vol. 59, pp. 892–902. https://doi.org/10.1373/clinchem.2013.206375
Salipante, S.J. and Jerome, K.R., Clin. Chem., 2020, vol. 66, pp. 117–123. https://doi.org/10.1373/clinchem.2019.304048
Khorosheva, E.M., Karymov, M.A., Selck, D.A., and Ismagilov, R.F., Nucleic Acids Res., 2016, vol. 44, p. e10. https://doi.org/10.1093/nar/gkv877
Schuler, F., Siber, C., Hin, S., Wadle, S., Paust, N., Zengerle, R., and von Stetten, F., Anal. Methods, 2016, vol. 8, pp. 2750–2755. https://doi.org/10.1039/C6AY00600K
Wong, Y.-P., Othman, S., Lau, Y.-L., Radu, S., and Chee, H.-Y., J. Appl. Microbiol., 2018, vol. 124, pp. 626–643. https://doi.org/10.1111/jam.13647
Land, K.J., Boeras, D.I., Chen, X.-S., Ramsay, A.R., and Peeling, R.W., Nat. Microbiol., 2019, vol. 4, pp. 46–54. https://doi.org/10.1038/s41564-018-0295-3
Coole, J., Kortum, A., Tang, Y., Vohra, I., Maker, Y., Kundrod, K., Natoli, M., and Richards-Kortum, R., J. Vis. Ex., vol. 168, pp. 1–14. https://doi.org/10.3791/62148
Mezin, A.V., Naumov, S.A., Prusakov, K.A., Nazarenko, K.A., Aldarov, K.G., Bazhutov, M.N., Naumov, A.Yu., Il’ina, E.N., Govorun, V.M., and Basmanov D.V., Patent RU 210215 U1, 2022.
Phillips, E.A., Moehling, T.J., Ejendal, K.F.K., Hoilett, O.S., Byers, K.M., Basing, L.A., Jankowski, L.A., Bennett, J.B., Lin, L.-K., Stanciu, L.A., and Linnes, J.C., Lab Chip, vol. 19, pp. 3375–3386. https://doi.org/10.1039/c9lc00506d
Priye, A., Bird, S.W., Light, Y.K., Ball, C.S., Negrete, O.A., and Meagher, R.J., Sci. Rep., vol. 7, p. 44778. https://doi.org/10.1038/srep44778
Fu, Y., Duan, X., Huang, J., Huang, L., Zhang, L., Cheng, W., Ding, S., and Min, X., Sci. Rep., vol. 9, p. 5955. https://doi.org/10.1038/s41598-019-42542-x
Ding, X., Wang, G., and Mu, Y., Anal. Chim. Acta, 2019, vol. 1081, pp. 193–199. https://doi.org/10.1016/j.aca.2019.07.055
Mitani, Y., Lezhava, A., Kawai, Y., Kikuchi, T., Oguchi-Katayama, A., Kogo, Y., Itoh, M., Miyagi, T., Takakura, H., Hoshi, K., Kato, C., Arakawa, T., Shibata, K., Fukui, K., Masui, R., Kuramitsu, S., Kiyotani, K., Chalk, A., Tsunekawa, K., Murakami, M., Kamataki, T., Oka, T., Shimada, H., Cizdziel, P.E., and Hayashizaki, Y., Nat. Methods, 2007, vol. 4, pp. 257–262. https://doi.org/10.1038/nmeth1007
Itonaga, M., Matsuzaki, I., Warigaya, K., Tamura, T., Shimizu, Y., Fujimoto, M., Kojima, F., Ichinose, M., and Murata, S.-I., PLoS One, 2016, vol. 11, p. e0151654. https://doi.org/10.1371/journal.pone.0151654
Cao, G., Kong, J., Xing, Z., Tang, Y., Zhang, X., Xu, X., Kang, Z., Fang, X., and Guan, M., Anal. Chim. Acta, 2018, vol. 1024, pp. 123–135. https://doi.org/10.1016/j.aca.2018.04.022
Paul, R., Ostermann, E., and Wei, Q., Biosens. Bioelectron., 2020, vol. 169, p. 112592. https://doi.org/10.1016/j.bios.2020.112592
Mason, M.G. and Botella, J.R., Nat. Protoc., 2020, vol. 15, pp. 3663–3677. https://doi.org/10.1038/s41596-020-0392-7
Nagura-Ikeda, M., Imai, K., Tabata, S., Miyoshi, K., Murahara, N., Mizuno, T., Horiuchi, M., Kato, K., Imoto, Y., Iwata, M., Mimura, S., Ito, T., Tamura, K., and Kato, Y., J. Clin. Microbiol., 2020, vol. 58, p. e01438-20. https://doi.org/10.1128/jcm.01438-20
Fakheran, O., Dehghannejad, M., and Khademi, A., Infect. Dis. Poverty, 2020, vol. 9, p. 100. https://doi.org/10.1186/s40249-020-00728-w
Kellner, M.J., Koob, J.G., Gootenberg, J.S., Abudayyeh, O.O., and Zhang, F., Nat. Protoc., 2019, vol. 14, pp. 2986–3012. https://doi.org/10.1038/s41596-019-0210-2
Gootenberg, J.S., Abudayyeh, O.O., Lee, J.W., Essletzbichler, P., Dy, A.J., Joung, J., Verdine, V., Donghia, N., Daringer, N.M., Freije, C.A., Myhrvold, C., Bhattacharyya, R.P., Livny, J., Regev, A., Koonin, E.V., Hung, D.T., Sabeti, P.C., Collins, J.J., and Zhang, F., Science, 2017, vol. 356, pp. 438–442. https://doi.org/10.1126/science.aam9321
Li, S.-Y., Cheng, Q.-X., Wang, J.-M., Li, X.-Y., Zhang, Z.-L., Gao, S., Cao, R.-B., Zhao, G.-P., and Wang, J., Cell Discov., 2018, vol. 4, p. 20. https://doi.org/10.1038/s41421-018-0028-z
Chen, J.S., Ma, E., Harrington, L.B., Da Costa, M., Tian, X., Palefsky, J.M., and Doudna, J.A., Science, 2018, vol. 360, pp. 436–439. https://doi.org/10.1126/science.aar6245
Butler, D., Mozsary, C., Meydan, C., Foox, J., Rosiene, J., Shaiber, A., Danko, D., Afshinnekoo, E., MacKay, M., Sedlazeck, F.J., Ivanov, N.A., et al., Nat. Commun., 2021, vol. 12, p. 1660. https://doi.org/10.1038/s41467-021-21361-7
Vandenberg, O., Martiny, D., Rochas, O., van Belkum, A., and Kozlakidis, Z., Nat. Rev. Microbiol., 2021, vol. 19, pp. 171–183. https://doi.org/10.1038/s41579-020-00461-z
ACKNOWLEDGMENTS
Authors are grateful to K.A. Miroshnikov, A.N. Ignatov, and A.M. Chuenko for sharing the collection of strains of phytopathogenic bacteria and the equipment of the PhytoEngineering research center. The work is dedicated to the memory of academician Yu.A. Ovchinnikov.
Funding
The work was supported by the Russian Science Foundation, project no. 20-75-10144.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
The paper does not contain a description of studies performed with the participation of people or animals as objects.
Additional information
Translated by N. Onishchenko
Abbreviations: Bst LF, large fragment of DNA polymerase I of Geobacillus stearothermophilus; LAMP, loop-mediated isothermal amplification; Taq polymerase, DNA polymerase I of Thermus aquaticus.
Rights and permissions
About this article

Cite this article
Shirshikov, F.V., Bespyatykh, J.A. Loop-Mediated Isothermal Amplification: From Theory to Practice. Russ J Bioorg Chem 48, 1159–1174 (2022). https://doi.org/10.1134/S106816202206022X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S106816202206022X