
Abstract
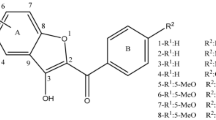
DNA binding position and binding affinity of drugs are important information that helps medicinal chemists in synthesis of new drugs. We used molecular docking and molecular dynamics simulation to reveal binding strength of thieno[2,3-b]benzo[1,8]naphthyridine derivatives to DNA. Molecular docking showed that molecules with more steric hindrance select groove position in DNA structure. Other molecules are intercalated between base pairs of GC and AT. Restrained electrostatic potential (RESP) charges, root mean square deviation (RMSD), and total potential analyses were performed. RMSD and total potential analyses showed that all simulations have stability for MMGBSA analysis. Binding affinity of all drugs was derived via MMGBSA analysis. Thermodynamics analysis showed that binding affinity of groove binding drugs is less than that of intercalating ones. Also, it was found that a linear relationship exists between RESP charges and ΔG pred. Additionally, our results demonstrated the highest affinity for molecules carrying substituent groups of–OCH3 and–CH3.
Similar content being viewed by others

References
Fadda, A.A., El-Defrawy, A.M., and El-Habiby, S.A., Am. J. Org. Chem., 2012, vol. 2, pp. 87–96.
Roma, G., Grossi, G., di Braccio, M., et al., Eur. J. Med. Chem., 2008, vol. 43, pp. 1665–1680.
Quintela, J.M., Peinador, C., González, L., et al., Eur. J. Med. Chem., 2003, vol. 38, pp. 265–275.
Aboul-Fadl, T., Bin-Jubair, F.A.S., and Aboul-Wafa O., Eur. J. Med. Chem., 2010, vol. 45, no. 10, pp. 4578–4586.
Baez, A., González, F.A., Vázquez, D., and Waring, M.J., Biochem. Pharmacol., 1983, vol. 32, pp. 2089–2094.
Cao, Y. and He, W.X., Spectrochim. Acta, 1998, pp. 883.
Singh, M.P., Joseph, T., Kumar, S., and Lown, J.W., Chem. Res. Toxicol., 1992, vol. 5, p. 597.
TilakRaj, T. and Ambekar, S.Y., J. Prakt. Chem., 1988, vol. 330, pp. 293–298.
Naik, T.R.R., Naik, H.S.B., Raghavedra M., and Naik, S.G.K., Arkivoc, 2006, vol. 2006, pp. 84–94.
Becke, A.D., J. Chem. Phys., 1993, vol. 98, pp. 5648–5652.
Frisch, M.J., Trucks, G.W., Schlegel H.B., et al., GAUSSIAN 03, Revision C.02 Gaussian, Inc., Wallingford CT, 2004.
Bayly, C.I., Cieplak, P., Cornell, W., and Kollman, P.A., J. Phys. Chem., 1993, vol. 97, pp. 10269–10280.
Mark, A., Argus Lab 4.0.1, Thompson, Planaria Software LLC, Seattle, WA. http://www.arguslab.com/.
Skauge, T., Turel, I., Sletten, E., Inorg. Chim. Acta., 2002, vol. 339, pp. 239–247.
Cheatham, T.E., Cieplak, P., and Kollman, P.A., J. Biomol. Struct. Dyn., 1999, vol. 16, pp. 845–862.
Pérez, A., Marchán, I., and Svozil, D., Biophys. J., 2007, vol. 92, pp. 3817–3829.
Wang, J., Wolf, R.M., Caldwell, J.W., Kollman, P.A., and Case, D.A., J. Comput. Chem., 2004, vol. 25, p. 1157–1174.
Berendsen, H.J.C., Postma, J.P.M., van Gunsteren, W.F., and Hermans, J., Intermolecular Forces, Pullman, B., Ed., Netherlands, 1981.
Parisi, G. and Yong-shi, Wu, Sci. Sin., 1981, vol. 24, pp. 483–496.
Kollman, P.A., Massova, I., and Reyes, C., Acc. Chem. Res., 2000, vol.33, pp.889–897.
Wang, J., Hou, T., and Xu, X., Curr. Comput.-Aided Drug Des., 2006, vol.2, pp.287–306.
Srinivasan, J., Cheatham, T.E., Cieplak, P., Kollman, P.A., and Case, D.A., J. Am. Chem. Soc., 1998, vol. 120, pp. 9401–9409.
Srinivasan, J., Miller, J., Kollman, P.A., and Case, D.A., J. Biomol. Struct. Dyn., 1998, vol.16, pp. 671–682.
Cheatham, T.E., Srinivasan, J., Case, D.A., and Kollman, P.A., J. Biomol. Struct. Dyn., 1998, vol. 16, pp. 265–280.
Chong, L.T., Duan, Y., Wang, L., Massova, I., and Kollman, P.A., Proc. Natl. Acad. Sci. U. S. A., 1999, vol. 96, pp. 14330–14335.
Reyes, C.M. and Kollman, P.A.,? J. Mol. Biol., 2000, vol. 297, pp. 1145–1158.
Nandeshwarappa, B.P., Aruna Kumar, D.B., Bhojya Naik, H.S., and Mahadevan, K.M., Phosphorus, Sulfu,r Silicon Relat. Elem., 2006, vol. 181, pp. 1997–2003.
Scatchard, G., Ann. N.Y. Acad. Sci., 1949, vol. 51, pp. 660–672.
Peacocke, A.R. and Skerrett, J.N.H., Trans. Faraday Soc.,1956, vol. 52, pp. 261–279.
Naik, T.R.R., Naik, H.S.B., Naik, H.R.P., Bindu, P.J., Harish, B.G., and Krishna, V., Med. Chem., 2009, vol. 5, pp. 411–418.
Author information
Authors and Affiliations
Corresponding author
Additional information
The article is published in the original.
Rights and permissions
About this article

Cite this article
Sargolzaei, M., Afshar, M. & Nikoofard, H. Molecular dynamics simulation study of binding affinity of thieno[2,3-b]benzo[1,8]naphthyridine derivatives to DNA. Russ J Bioorg Chem 43, 435–442 (2017). https://doi.org/10.1134/S1068162017040057
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162017040057