
Abstract
Polyglutamine (polyQ) disorders are severe forms of inherited neurodegenerative pathologies caused by the expansion of CAG repeats in specific genes accompanied by abnormal elongation of the polyglutamic residues in the mutant part of the protein, impaired protein-protein interactions, and formation of insoluble amyloid inclusions in neurons. Therapy for these diseases is only symptomatic, and it cannot affect the dynamics of the pathological process. To date, nine polyQ diseases are known: spinocerebellar ataxia type 1, 2, 3, 6, 7, and 17, spino-bulbar muscular atrophy, dentatorubral-pallidoluysian atrophy, and Huntington’s disease. Expansion of trinucleotide repeats can take place during processes of replication, transcription and reparation, though the exact mechanisms are still unclear. There are many ways for CAG expansion resulting in neurodegeneration. Nearly complete penetrance of mutant alleles, severe disabilities after 10–15 years from the disease onset, and lack of effective therapy contribute to consideration of polyQ disorders as a very significant problem actively investigated worldwide. This review is focused on the existing models and molecular mechanisms of spinocerebellar ataxia type 1 (SCA1), one of the polyglutamine disorders, which is caused by expansion of CAG repeats in the ATXN1 gene.

Similar content being viewed by others

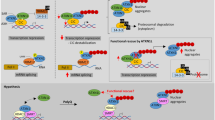
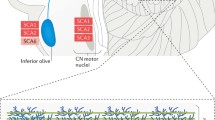
REFERENCES
Illarioshkin, S.N., Rudenskaya, G.E., Ivanova-Smolenskaya, I.A., et al., Nasledstvennye ataksii i paraplegii (Hereditary Ataxia and Paraplegia), Moscow: MEDpress-Inform, 2006.
Stoyas, C. and La Spada, A., The CAG-polyglutamine repeat diseases: a clinical, molecular, genetic, and pathophysiologic nosology, Handb. Clin. Neurol., 2018, vol. 147, pp. 143—170https://doi.org/10.1016/B978-0-444-63233-3.00011-7
Katsuno, M., Tanaka, F., Adachi, H., et al., Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA), Prog. Neurobiol., 2012, vol. 99, no. 3, pp. 246—256. https://doi.org/10.1016/j.pneurobio.2012.05.007
Ha, A.D. and Fung, V.S., Huntington’s disease, Curr. Opin. Neurol., 2012, vol. 25, no. 4, pp. 491—498. https://doi.org/10.1097/WCO.0b013e3283550c97
Storey, E., Genetic cerebellar ataxias, Semin. Neurol., 2014, vol. 34, no. 3, pp. 280—292. https://doi.org/10.1055/s-0034-1386766
Stevanin, G. and Brice, A., Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4), Cerebellum, 2008, vol. 7, no. 2, pp. 170—178. https://doi.org/10.1007/s12311-008-0016-1
Zoghbi, H.Y. and Orr, H.T., Glutamine repeats and neurodegeneration, Annu. Rev. Neurosci., 2000, vol. 23, pp. 217—247. https://doi.org/10.1146/annurev.neuro.23.1.217
Sun, Y.M., Zhang, Y.B., and Wu, Z.Y., Huntington’s disease: relationship between phenotype and genotype, Mol. Neurobiol., 2017, vol. 54, no. 1, pp. 342—348. https://doi.org/10.1007/s12035-015-9662-8
Schut, J., Hereditary ataxia: clinical study through six generations, Arch.NeurPsych., 1950, vol. 63, pp. 535—568.
Koneva, L.A., Kucher, A.N., Puzyrev, V.P., et al., Demographic and clinical-genetic features of the type I spinocerebellar ataxia prevalence among the Ust-Aldan and Aby Uluses in the Republic of Sakha (Yakutia), Aktual’nye voprosy profilakticheskoi meditsiny (Current Issues in Preventive Medicine) (Proc. Theor. Pract. Cof.), Ulan-Ude, 2005, pp. 97—100.
Banfi, S., Servadio, A., Chung, M., et al., Identification and characterization of the gene causing type 1 spinocerebellar ataxia, Nat. Genet., 1994, vol. 7, pp. 513—520. https://doi.org/10.1038/ng0894-513
Opal, P. and Ashizawa, T., Spinocerebellar ataxia type 1, GeneReviews (Internet), 1998.
Kraus-Perrotta, C. and Lagalwar, S., Expansion, mosaicism and interruption: mechanisms of the CAG repeat mutation in spinocerebellar ataxia type 1, Cerebellum Ataxias, 2016, vol. 3, p. 20. https://doi.org/10.1186/s40673-016-0058-y
Pearson, C., Eichler, E., Lorenzetti, D., et al., Interruptions in the triplet repeats of SCA1 and FRAXA reduce the propensity and complexity of slipped strand DNA (S-DNA) formation, Biochemistry, 1998, vol. 37, no. 8, pp. 2701—2708. https://doi.org/10.1021/bi972546c
Matilla-Dueñas, A., Goold, R., and Giunti, P., Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1, Cerebellum, 2008, pp. 106—114. https://doi.org/10.1007/s12311-008-0009-0
Seidel, K., Siswanto, S., Ewout, R., et al., Brain pathology of spinocerebellar ataxias, Acta Neuropathol., 2012, vol. 124, pp. 1—21. https://doi.org/10.1007/s00401-012-1000-x
Biirk, K., Abele, M., Fetter, M., et al., Autosomal dominant cerebellar ataxia type I: clinical features and MRI in families with SCA1, SCA2 and SCA3, Brain, 1996, vol. 119, pp. 1497—1505. https://doi.org/10.1093/brain/119.5.1497
Rüb, U., Bürk, K., Timmann, D., et al., Spinocerebellar ataxia type 1 (SCA1): new pathoanatomical and clinico-pathological insights, Neuropathol. Appl. Neurobiol., 2012, vol. 38, pp. 665—680. https://doi.org/10.1111/j.1365-2990.2012.01259.x
Guerrini, L., Lolli, F., Ginestroni, A., et al., Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not in SCA2: a quantitative volumetric, diffusion and proton spectroscopy MR study, Brain, 2004, vol. 127, pp. 1785—1795. https://doi.org/10.1093/brain/awh201
Adanyeguh, I., Henry, P., Nguyen, T. et al., In vivo neurometabolic profiling in patients with spinocerebellar ataxia types 1, 2, 3 and 7, Mov. Disord., 2015, vol. 30, no. 5, pp. 662—670. https://doi.org/10.1002/mds.26181
Burright, E., Clark, H., Servadio, A., et al., SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat, Cell, 1995, vol. 82, pp. 937—948. https://doi.org/10.1016/0092-8674(95)90273-2
Matilla, A., Roberson, D., Banfi, S., et al., Mice lacking ataxin-1 display learning deficits and decreased hippocampal paired-pulse facilitation, J. Neurosci., 1998, vol. 18, pp. 5508—5516. https://doi.org/10.1523/JNEUROSCI.18-14-05508.1998
Watase, K., Weeber, E., Xu, B., et al., A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration, Neuron, 2002, vol. 34, pp. 905—919. https://doi.org/10.1016/S0896-6273(02)00733-X
Lorenzetti, D., Watase, K., Xu, B., et al., Repeat instability and motor incoordination in mice with a targeted expanded CAG repeat in the Sca1 locus, Hum. Mol. Genet., 2000, vol. 9, pp. 779—785. https://doi.org/10.1093/hmg/9.5.779
Zu, T., Duvick, L., Kaytor, M., et al., Recovery from polyglutamine-induced neurodegeneration in conditional SCA1 transgenic mice, J. Neurosci., 2004, vol. 24, pp. 8853—8861. https://doi.org/10.1523/JNEUROSCI.2978-04.2004
Giovannoni, R., Maggio, N., Rosaria-Bianco, M., et al., Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model, Neuron Glia Biol., 2007, vol. 3, no. 4, pp. 335—351. https://doi.org/10.1017/S1740925X08000185
Ingram, M., Orr, H., and Clark, H., Genetically engineered mouse models of the trinucleotide-repeat spinocerebellar ataxias, Brain Res. Bull., 2012, vol. 88, no. 1, pp. 33—42. https://doi.org/10.1016/j.brainresbull.2011.07.016
Emamian, E.S., Kaytor, M.D., Duvick, L.A., et al., Serine 776 of ataxin-1 is critical for polyglutamine induced disease in SCA1 transgenic mice, Neuron, 2003, vol. 38, pp. 375—387. https://doi.org/10.1016/S0896-6273(03)00258-7
Duvick, L., Barnes, J., Ebner, B., et al., SCA1-like disease in mice expressing wild-type ataxin-1 with a serine to aspartic acid replacement at residue 776, Neuron, 2010, vol. 67, pp. 929—935. https://doi.org/10.1016/j.neuron.2010.08.022
Fernandez-Funez, P., Nino-Rosales, M.L., de Gouyon, B., et al., Identification of genes that modify ataxin-1-induced neurodegeneration, Nature, 2000, vol. 408, no. 6808, pp. 101—106. https://doi.org/10.1038/35040584
Bondar, V.V., Adamski, C.J., Onur, T.S., et al., PAK1 regulates ATXN1 levels providing an opportunity to modify its toxicity in spinocerebellar ataxia type 1, Hum. Mol. Genet., 2018, vol. 27, no. 16, pp. 2863—2873. https://doi.org/10.1093/hmg/ddy200
Barclay, S.S., Tamura, T., Ito, H., et al., Systems biology analysis of Drosophila in vivo screen data elucidates core networks for DNA damage repair in SCA1, Hum. Mol. Genet., 2014, vol. 23, no. 5, pp. 1345—1364. https://doi.org/10.1093/hmg/ddt524
Skinner, P.J., Koshy, B.T., Cummings, C.J., et al., Ataxin-1 with an expanded glutamine tract alters nuclear matrix-associated structures, Nature, 1997, vol. 391, no. 6664, pp. 307—307. https://doi.org/10.1038/34701
Krol, H.A., Krawczyk, P.M., Bosch, K.S., et al., Polyglutamine expansion accelerates the dynamics of ataxin-1 and does not result in aggregate formation, PLoS One, 2008, vol. 3, no. 1. e1503. https://doi.org/10.1371/journal.pone.0001503
Mapelli, L., Canale, C., Pesci, D., et al., Toxic effects of expanded ataxin-1 involve mechanical instability of the nuclear membrane, Biochim. Biophys. Acta,Mol. Basis Dis., 2012, vol. 1822, no. 6, pp. 906—917. https://doi.org/10.1016/j.bbadis.2012.01.016
Lee, Y., Samaco, R.C., Gatchel, J.R., et al., miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis, Nat. Neurosci., 2008, vol. 11, no. 10, pp. 1137—1139. https://doi.org/10.1038/nn.2183
Frattini, A., Fabbri, M., Valli, R., et al., High variability of genomic instability and gene expression profiling in different HeLa clones, Sci. Rep., 2015, vol. 5, no. 1. https://doi.org/10.1038/srep15377
Lin, Y.-C., Boone, M., Meuris, L., et al., Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations, Nat. Comm., 2014, vol. 1. https://doi.org/10.1038/ncomms5767
Takahashi, K. and Yamanaka, S., Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors, Cell, 2006, vol. 126, no. 4, pp. 663—676. https://doi.org/10.1016/j.cell.2006.07.024
Muguruma, K., Nishiyama, A., Kawakami, H., et al., Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells, Cell Rep., 2015, vol. 10, no. 4, pp. 537—550. https://doi.org/10.1016/j.celre.2014.12.051
Qian, X., Jacob, F., Song, M.M., et al., Generation of human brain region-specific organoids using a miniaturized spinning bioreactor, Nat. Protoc., 2018, vol. 13, no. 3, pp. 565—580. https://doi.org/10.1038/nprot.2017.152
Gorris, R., Fischer, J., Erwes, K.L., et al., Pluripotent stem cell-derived radial glia-like cells as stable intermediate for efficient generation of human oligodendrocytes, Glia, 2015, vol. 63, no. 12, pp. 2152—2167. https://doi.org/10.1002/glia.22882
Douvaras, P., Sun, B., Wang, M., et al., Directed differentiation of human pluripotent stem cells to microglia, Stem Cell Rep., 2017, vol. 8, no. 6, pp. 1516—1524. https://doi.org/10.1016/j.stemcr.2017.04.023
Lebedeva, O.S., Surdina, A.V., Bogomiakova, M.E., et al., IPSC-based model for the study of molecular mechanisms of spinocerebellar ataxia type 1, FEBS J., 2017, vol. 284, suppl. 1, p. 141.
Rousseaux, M.W.C., Tschumperlin, T., Lu, H.-C., et al., ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism, Neuron, 2018, vol. 97, no. 6, pp. 1235—1243. https://doi.org/10.1016/j.neuron.2018.02.013 45a. Bogomazova, A.N., Vasina, E.M., Kiselev, S.L., et al., Genetic cell reprogramming: a new technology for basic research and applied usage, Russ. J. Genet., 2015, vol. 51, no. 4, pp. 386—396. https://doi.org/10.1134/S102279541504002X
Serra, H.G., Byam, C.E., Lande, J.D., et al., Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice, Hum. Mol. Genet., 2004, vol. 13, no. 20, pp. 2535—2543. https://doi.org/10.1093/hmg/ddh268
Notartomaso, S., Zapulla, C., Biagioni, F., et al., Pharmacological enhancement of mGlu1 metabotropic glutamate receptors causes a prolonged symptomatic benefit in a mouse model of spinocerebellar ataxia type 1, Mol. Brain, 2013, vol. 6, no. 48. https://doi.org/10.1186/1756-6606-6-48
Power, E.M., Morales, A., and Empson, R.M., Prolonged type 1 metabotropic glutamate receptor dependent synaptic signaling contributes to spino-cerebellar ataxia type 1, J. Neurosci., 2016, vol. 36, no. 18, pp. 4910—4916. https://doi.org/10.1523/jneurosci.3953-15.2016
Shuvaev, A.N., Hosoi, N., Sato, Y., et al., Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice, J. Physiol., 2016, vol. 595, no. 1, pp. 141—164. https://doi.org/10.1113/jp272950
Oz, G., Kittelson, E., Demirgöz, D., et al., Assessing recovery from neurodegeneration in spinocerebellar ataxia: 1. Comparison of in vivo magnetic resonance spectroscopy with motor testing, gene expression and histology, Neurobiol. Dis., 2015, vol. 74, pp. 158—166. https://doi.org/10.1016/j.nbd.2014.11.011
Hourez, R., Servais, L., Orduz, D., et al., Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1, J. Neurosci., 2011, vol. 31, no. 33, pp. 11795—11807. https://doi.org/10.1523/jneurosci.0905-11.2011
Cvetanovic, M., Hu, Y.-S., and Opal, P., Mutant ataxin-1 inhibits neural progenitor cell proliferation in SCA1, Cerebellum, 2017, vol. 16, no. 2, pp. 340—347. https://doi.org/10.1007/s12311-016-0794-9
Cvetanovic, M., Decreased expression of glutamate transporter GLAST in Bergmann glia is associated with the loss of Purkinje neurons in the spinocerebellar ataxia type 1, Cerebellum, 2014, vol. 14, no. 1, pp. 8—11. https://doi.org/10.1007/s12311-014-0605-0
Ito, H., Fujita, K., Tagawa, K., et al., HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice, EMBO Mol. Med., 2014, vol. 7, no. 1, pp. 78—101. https://doi.org/10.15252/emmm.201404392
Taniguchi, J.B., Kondo, K., Fujita, K., et al., RpA1 ameliorates symptoms of mutant ataxin-1 knock-in mice and enhances DNA damage repair, Hum. Mol. Genet., 2016, vol. 25, no. 20, pp. 4432—4447. https://doi.org/10.1093/hmg/ddw272
Qi, M.-L., Tagawa, K., Enokido, Y., et al., Proteome analysis of soluble nuclear proteins reveals that HMGB1/2 suppress genotoxic stress in polyglutamine diseases, Nat. Cell Biol., 2007, vol. 9, no. 4, pp. 402—414. https://doi.org/10.1038/ncb1553
Ryu, J. and Lee, D.H., Dual-specificity phosphatase 18 modulates the SUMOylation and aggregation of ataxin-1, Biochem. Biophys. Res. Commun., 2018, vol. 502, no. 3, pp. 389—396. https://doi.org/10.1016/j.bbrc.2018.05.178
Cvetanovic, M., Ingram, M., Orr, H., and Opal, P., Early activation of microglia and astrocytes in mouse models of spinocerebellar ataxia type 1, Neuroscience, 2015, vol. 289, pp. 289—299. https://doi.org/10.1016/j.neuroscience.2015.01.003
Rodriguez-Lebron, E., Liu, G., Keiser, M., et al., Altered Purkinje cell miRNA expression and SCA1 pathogenesis, Neurobiol. Dis., 2013, vol. 54, pp. 456—463. https://doi.org/10.1016/j.nbd.2013.01.019
Cummings, C.J., Reinstein, E., Sun, Y., et al., Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice, Neuron, 1999, vol. 24, no. 4, pp. 879—892. https://doi.org/10.1016/s0896-6273(00)81035-1
Stenoien, D.L., Mielke, M., and Mancini, M.A., Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components, Nat. Cell Biol., 2002, vol. 4, no. 10, pp. 806—810. https://doi.org/10.1038/ncb859
Cummings, C.J., Mancini, M.A., Antalffy, B., et al., Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1, Nat. Genet., 1998, vol. 19, no. 2, pp. 148—154. https://doi.org/10.1038/502
Vig, P., Hearst, S., Shao, Q., et al., Glial S100B protein modulates mutant ataxin-1 aggregation and toxicity: TRTK12 peptide, a potential candidate for SCA1 therapy, Cerebellum, 2011, vol. 10, no. 2, pp. 254—266. https://doi.org/10.1007/s12311-011-0262-5
Serra, H.G., Duvick, L., Zu, T., et al., RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice, Cell, 2006, vol. 127, no. 4, pp. 697—708. https://doi.org/10.1016/j.cell.2006.09.036
Tsuda, H., Jafar-Nejad, H., Patel, A.J., et al., The AXH domain of ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins, Cell, 2005, vol. 122, no. 4, pp. 633—644. https://doi.org/10.1016/j.cell.2005.06.012
Mizutani, A., Wang, L., Rajan, H., et al., Boat, an AXH domain protein, suppresses the cytotoxicity of mutant ataxin-1, EMBO J., 2005, vol. 24, no. 18, pp. 3339—3351. https://doi.org/10.1038/sj.emboj.7600785
Yue, S., Serra, S., Zoghbi, H., and Orr, H., The spinocerebellar ataxia type 1 protein, ataxin-1, has RNA-binding activity that is inversely affected by the length of its polyglutamine tract, Hum. Mol. Genet., 2001, vol. 10, no. 1, pp. 25—30. https://doi.org/10.1093/hmg/10.1.25
De Chiara, C., Menon, R.P., Dal Piaz, F., et al., Polyglutamine is not all: the functional role of the AXH domain in the ataxin-1 protein, J. Mol. Biol., 2005, vol. 354, no. 4, pp. 883—893. https://doi.org/10.1016/j.jmb.2005.09.083
Irwin, S., Vandelft, M., Pinchev, D., et al., RNA association and nucleocytoplasmic shuttling by ataxin-1, J. Cell Sci., 2005, vol. 118, no. 1, pp. 233—242. https://doi.org/10.1242/jcs.01611
Lam, Y.C., Bowman, A.B., Jafar-Nejad, P., et al., ATAXIN-1 interacts with the repressor capicua in its native complex to cause SCA1 neuropathology, Cell, 2006, vol. 127, no. 7, pp. 1335—1347. https://doi.org/10.1016/j.cell.2006.11.038
Crespo-Barreto, J., Fryer, J.D., Shaw, C.A., et al., Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis, PLoS Genet., 2010, vol. 6, no. 7. e1001021. https://doi.org/10.1371/journal.pgen.1001021
Sánchez, I., Balagué, E., and Matilla-Dueñas, A., Ataxin-1 regulates the cerebellar bioenergetics proteome through the GSK3β-mTOR pathway which is altered in spinocerebellar ataxia type 1 (SCA1), Hum. Mol. Genet., 2016, vol. 25, no. 18, pp. 4021—4040. https://doi.org/10.1093/hmg/ddw242
Davidson, J.D., Riley, B., Burright, E.N., et al., Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein, Hum. Mol. Genet., 2000, vol. 9, no. 15, pp. 2305—2312. https://doi.org/10.1093/oxfordjournals.hmg.a018922
Okazawa, H., Rich, T., Chang, A., et al., Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death, Neuron, 2002, vol. 34, no. 5, pp. 701—713.https://doi.org/10.1016/s0896-6273(02)00697-9
Hong, S., Kim, S.-J., Ka, S., et al., USP7, a ubiquitin-specific protease, interacts with ataxin-1, the SCA1 gene product, Mol. Cell. Neurosci., 2002, vol. 20, no. 2, pp. 298—306. https://doi.org/10.1006/mcne.2002.1103
Goold, R., Hubank, M., Hunt, A., et al., Down-regulation of the dopamine receptor D2 in mice lacking ataxin 1, Hum. Mol. Genet., 2007, vol. 16, no. 17, pp. 2122—2134. https://doi.org/10.1093/hmg/ddm162
Tong, X., Gui, H., Jin, F., et al., Ataxin-1 and brother of ataxin-1 are components of the notch signalling pathway, EMBO Rep., 2011, vol. 12, no. 5, pp. 428—435. https://doi.org/10.1038/embor.2011.49
Klement, I.A., Skinner, P.J., Kaytor, M.D., et al., Ataxin-1 nuclear localization and aggregation, Cell, 1998, vol. 95, no. 1, pp. 41—53. https://doi.org/10.1016/s0092-8674(00)81781-x
Bañez-Coronel, M., Ayhan, F., Tarabochia, A.D., et al., RAN translation in Huntington disease, Neuron, 2015, vol. 88, pp. 667—677. https://doi.org/10.1016/j.neuron.2015.10.038
Zu, T., Gibbens, B., Doty, N.S., et al., Non-ATG-initiated translation directed by microsatellite expansions, Proc. Natl. Acad. Sci. U.S.A., 2010, vol. 108, no. 1, pp. 260—265. https://doi.org/10.1073/pnas.1013343108
Luján, R., Nusser, Z., Roberts, J.D.B., et al., Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus, Eur. J. Neurosci., 1996, vol. 8, no. 7, pp. 1488—1500. https://doi.org/10.1111/j.1460-9568.1996.tb01611.x
Lehre, K.P. and Danbolt, N.C., The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain, J. Neurosci., 1998, vol. 18, pp. 8751—8757. https://doi.org/10.1523/JNEUROSCI.18-21-08751.1998
Rothstein, J.D., Martin, L., Levey, A.I., et al., Localization of neuronal and glial glutamate transporters, Neuron, 1994, vol. 13, pp. 713—725. https://doi.org/10.1016/0896-6273(94)90038-8
Tanaka, J., Ichikawa, R., Watanabe, M., et al., Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses, NeuroReport, 1997, vol. 8, no. 11, pp. 2461—2464. https://doi.org/10.1097/00001756-199707280-00010
Furuta, A., Martin, L., Lin, C., et al., Cellular and synaptic localization of the neuronal glutamate transporters excitatory amino acid transporter 3 and 4, Neuroscience, 1997, vol. 81, no. 4, pp. 1031—1042. https://doi.org/10.1016/s0306-4522(97)00252-2
Havel, L.S., Li, S., and Li, X., Nuclear accumulation of polyglutamine disease proteins and neuropathology, Mol. Brain, 2009, vol. 2, no. 21. https://doi.org/10.1186/1756-6606-2-21
Nekrasov, E.D., Vigont, V.A., Klyushnikov, S.A., et al., Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons, Mol. Neurodegener., 2016, vol. 11, no. 1. https://doi.org/10.1186/s13024-016-0092-5
Gasset-Rosa, F., Chillon-Marinas, C., Goginashvili, A., et al., Polyglutamine-expanded Huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport, Neuron, 2017, vol. 94, no. 1, pp. 48—57. https://doi.org/10.1016/j.neuron.2017.03.027
Monoi, H., Futaki, S., Kugimiya, S., et al., Poly-l-glutamine forms cation channels: relevance to the pathogenesis of the polyglutamine diseases, Biophys. J., 2000, vol. 78, no. 6, pp. 2892—2899. https://doi.org/10.1016/s0006-3495(00)76830-5
Hirakura, Y., Azimov, R., Azimova, R., and Kagan, B.L., Polyglutamine-induced ion channels: a possible mechanism for the neurotoxicity of Huntington and other CAG repeat diseases, J. Neurosci. Res., 2000, vol. 60, no. 4, pp. 490—494. https://doi.org/10.1002/(SICI)1097-4547(20000515)60:4<490::AID-JNR7>3.0.CO;2-9
Cvetanovic, M., Patel, J.M., Marti, H.H., et al., Vascular endothelial growth factor ameliorates the ataxic phenotype in a mouse model of spinocerebellar ataxia type 1, Nat. Med., 2011, vol. 17, no. 11, pp. 1445—1447. https://doi.org/10.1038/nm.2494
Persengiev, S., Kondova, I., Otting, N., et al., Genome-wide analysis of miRNA expression reveals a potential role for miR-144 in brain aging and spinocerebellar ataxia pathogenesis, Neurobiol. Aging, 2011, vol. 32, no. 12, pp. 17—27. https://doi.org/10.1016/j.neurobiolaging.2010.03.014
Funding
The present study was supported by the Russian Science Foundation (project no. 19-15-00425).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by A. Kazantseva
Rights and permissions
About this article

Cite this article
Volovikov, E.A., Davidenko, A.V. & Lagarkova, M.A. Molecular Mechanisms of Spinocerebellar Ataxia Type 1. Russ J Genet 56, 129–141 (2020). https://doi.org/10.1134/S102279542002012X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S102279542002012X