
Abstract
A study is performed of the adsorption of water–methanol and water–acetonitrile mixtures on chiral stationary phases (CSPs) Chirobiotic R, Chirobiotic T, and Nautilus-E with grafted macrocyclic antibiotics ristocetin A, teicoplanin, and eremomycin, respectively. The patterns of adsorption on the indicated CSPs are qualitatively the same, and differ only by quantitative indicators. Adsorption isotherms of excess water from binary solvents have adsorption azeotrope points and show the preferred absorption of water in the range of pure organic component to an azeotrope point in the range of 60–75 mol % for H2О–МеОН and 80–90 mol % for H2O–MeCN systems. It is shown that the thickness of the adsorption phase in the first case is less than one nominal molecular layer (0.10–0.13 nm). For H2O–MeCN, it is 3–4 molecular layers (0.88–1.05 nm). Activity coefficients are calculated for the components of solutions in surface layers. The coefficients indicate the systems deviate considerably from the properties of an ideal adsorption solution. Reasons for this behavior are discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The structure of a phase boundary has a great effect on the mechanisms of retention and separation in liquid chromatography [1–3]. Systematic studies of the adsorption of binary solvents on chromatographic adsorbents show that the composition of the near-surface (adsorbed) layer of the solvent differs from that in the bulk of the liquid phase [1, 4–7]. The procedure for measuring the excess adsorption isotherms of components of binary mixtures is now used to study the properties of stationary phases [3, 8]. The objects of study in these works were silica gels and hydrophobized silica gels, which are typical adsorbents in normal-phase and reversed-phase chromatography, respectively. This technique is just beginning to gain popularity in chiral chromatography. It has been used to study the adsorption of binary eluents on chiral stationary phases (CSPs) Whelk-O1 [9], DACH-ACR [10], and Chiralcel OD-I [11]. The adsorption of aqueous organic solvents was used to study the surface properties of CSPs with grafted macrocyclic antibiotics in [12, 13], and the authors of [14] investigated the effect the composition of an adsorbed layer of the mobile phase has on the mechanism of amino acid retention on antibiotic columns. The attention researchers give to adsorbents with grafted antibiotics is due to their universal enantioselectivity and compatibility with both polar and non-polar solvents [15].
CSPs with a variety of antibiotics are used in analytical practice. It is therefore of interest to study the effect the nature of the selector has on the hydrophobic/hydrophilic properties of a stationary phase. In this work, we compare the adsorption of water–methanol and water–acetonitrile mixtures on CSPs with antibiotic-grafted eremomycin [12], ristocetin A, and teicoplanin. The adsorption of aqueous-organic mixtures on a CSP with the last antibiotic (Chirobiotic T) was studied in [13], but the adsorption values given in it were two orders of magnitude lower than the ones usually observed on polar CSPs. We therefore repeated the measurements on this adsorbent.
THEORETICAL
In this work, we used the definition of excess adsorption of component i (Gi) as its excess in a real system consisting of liquid and solid phases, relative to its content in a hypothetical reference system in which there is no adsorption interaction and the concentration of the component remains equal to its volume concentration at the interface [1]. The volume of the liquid phase in the column (dead volume, V0) remains unchanged in a chromatographic experiment, so we assumed the volumes of the solution in the real and reference systems were equal. This amount of excess adsorption was denoted as \(\Gamma _{i}^{{\left( v \right)}}\). It is easy to perform a theoretical analysis of adsorption equilibrium for a situation in which the numbers of moles of the liquid phase in the real system and the reference system are equal. This amount of excess adsorption is denoted as \(\Gamma _{i}^{{\left( n \right)}}\). In the adsorption of a binary solution, these two quantities are related by the simple relationship [1, 16]
where \(x_{i}^{l}\) denotes the mole fraction of component i in the liquid phase.
In the model of a layer of finite thickness [16], it is assumed that a hypothetical plane exists in the liquid phase at distance τ from the adsorbent surface, below which all of the excess of the adsorbed component is concentrated, and above which its concentration is equal to equilibrium concentration ci in the volume of the liquid phase. The value of τ is thus the thickness of the adsorption layer. The total amount of substance i in this layer (total adsorption) is
In this equation, total adsorption Q and excessive adsorption \({{\Gamma }^{{({v})}}}\) are expressed per unit surface area of the adsorbent. The value of τ can be estimated according to Schay and Nagy [17], based on the assumption that the linear section of the excess adsorption isotherm corresponds to the formation of a saturated adsorption layer of component i. In other words, dQi/dci = 0 and \(\tau = - (d\Gamma _{i}^{{\left( {v} \right)}}{\text{/}}d{{c}_{i}})\) in this section.
Intermolecular interaction in the adsorption layer can be analyzed on the basis of activity coefficients (γ) of components of the solution. The latter values in both the adsorption and the liquid phases are conveniently determined so that \(\gamma _{i}^{a}(\gamma _{i}^{l}) \to 1\) when \(x_{i}^{a}(x_{i}^{l}) \to 1\) (superscripts a and l refer to the adsorption and liquid phases, respectively). In [18], Larionov and Myers proposed an algorithm for calculating quantities γa that starts by calculating the logarithm of ratio \({{\gamma _{1}^{a}} \mathord{\left/ {\vphantom {{\gamma _{1}^{a}} {\gamma _{2}^{a}}}} \right. \kern-0em} {\gamma _{2}^{a}}}\):
Here, \(S = (x_{1}^{a}x_{2}^{l}){\text{/}}(x_{2}^{a}x_{1}^{l})\), where σ is the surface tension at the liquid/solid interface and \(\sigma _{i}^{0}\) is the surface tension of pure liquid i on the border with the solid. The molar fractions of the components of the solution in the adsorption layer, which are needed to calculate S, are found as \(x_{i}^{a} = {{{{Q}_{i}}} \mathord{\left/ {\vphantom {{{{Q}_{i}}} {\left( {{{Q}_{1}} + {{Q}_{2}}} \right)}}} \right. \kern-0em} {\left( {{{Q}_{1}} + {{Q}_{2}}} \right)}}\). To calculate the third and fourth terms on the right-hand side of the equation, we obtain the expressions
The activity coefficients for the liquid phase were calculated according to UNIQUAC [19] using model parameters taken from [20]: for the system water (1)–methanol (2), z = 10; r1 = 0.92; r2 = 1.4311; q1 = 1.40; q2 = 1.432; \(q_{1}^{'}\) = 1.00; \(q_{2}^{'}\) = 0.96; a12 = 289.6; and a21 = −181.0. For the system water (1)–acetonitrile (2), z = 10; r1 = 0.92; r2 = 1.8701; q1 = \(q_{1}^{'}\) = 1.40; q2 = \(q_{1}^{'}\) = 1.724; a12 = 112.6; and a21 = 242.8. The denotation of the parameters is the same as in [19]. Excess adsorption \(\Gamma _{i}^{{\left( n \right)}}\) were found from experimentally measured values of adsorption \(\Gamma _{i}^{{\left( {v} \right)}}\) for a given value \(x_{i}^{l}\) according to Eq. (1). Having determined ratio \({{\gamma _{1}^{a}} \mathord{\left/ {\vphantom {{\gamma _{1}^{a}} {\gamma _{2}^{a}}}} \right. \kern-0em} {\gamma _{2}^{a}}}\) according to Eq. (3), we calculated the activity coefficients for the surface layer as
The determined integrals in Eqs. (4) and (5) were found according to the trapezoidal rule.
EXPERIMENTAL
Equipment
Our experiments were performed on an LC20AD-XR liquid chromatograph (Shimadzu, Japan) equipped with a precision pump, refractometric detector, diode array detector (used only in experiments to determine the dead volume with 1,3,5-tri-tert-butylbenzene), autosampler, and column oven. The extracolumn volume measured in the system without a column that had a refractometric detector was 0.113 mL. In the system with a diode array detector, it was 0.054 mL. Corresponding amendments were considered when determining the retention volumes.
Reagents and Columns
We used Chirobiotic R (25 × 0.46 cm) and Chirobiotic T (25 × 0.46 cm) columns from Supelco (United States) packed with silica gel (particle size, 5 μm) with grafted antibiotics ristocetin A and teicoplanin, respectively. The packing weight in both columns was 2.5 g, and the specific surface area of the adsorbent was 300 m2/g (according to the manufacturer’s data).
Mobile phases were prepared from chemically pure methanol (Vekton, St. Petersburg), acetonitrile for HPLC (J.T. Baker, United States), and deionized water. Pure solvents were dried when used as the mobile phase: methanol, via distillation over magnesium methoxide [21]; acetonitrile, via distillation over zeolite 4A. In other cases, the solvents were used without additional purification. Deuterated methanol (99.8 at %; Acros, Belgium) was used to measure the dead volume isotopically.
Measuring Column Dead Volume
Column dead volume was measured in three ways: by the isotopic [22] and minor perturbation [23] methods, and according to the time of the release of an unretained tracer. In the first case, the dead volume was defined as the retention volume of deuterated methanol (2 μL) eluted with pure methanol. The dead volume was determined from minor perturbations according to Eq. (7), using data obtained by measuring the excess adsorption isotherms. When determining V0 with an unretained tracer, we used 1,3,5-tri-tert-butylbenzene (TtBB) with a concentration of 0.7 g/L and eluted with pure methanol. The sample volume was 2 μL. All measurements were made at a temperature of 25°C and a mobile phase flow rate of 1 mL/min.
Measuring Excess Adsorption Isotherms
Measurements were made for water–methanol and water–acetonitrile mixtures using minor disturbance method [6, 23] at a temperature of 25°C. Perturbation consisted of introducing a sample of a binary solvent into the column that differed slightly from the composition of the mobile phase. The measuring procedure was described in [12]. Our experiments were performed with mixtures that were 0, 0.5, 1, 2, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 95, 98, 99, 99.5, and 100 vol % organic solvent (MeOH or MeCN). The sample volume was 2 μL. The value of excess adsorption was found by numerically integrating the dependence of the retained volume of perturbation on the composition of the mobile phase, according to the equation
where A is the surface area of the stationary phase; VR is the retained volume of the perturbation at concentration plateau ci of component i; and VT is the thermodynamic dead volume, which is determined using the equation
where \(c_{i}^{0}\) is the pure liquid molar concentration of i.
RESULTS AND DISCUSSION
Dead Volume
Values of V0 found in different ways are compared in Table 1. The results obtained by the minor perturbation and isotopic methods are in good agreement with one another. The discrepancies between them are no more than 5% and are fully explained by the experimental errors. A considerable discrepancy of no less than 12% is observed between the value obtained isotopically and the retained volume of TtBB. The columns therefore have a small volume available for solvent molecules, but not for larger TtBB molecules. It is logical to assume that this volume lies in the space between the grafted selectors and the surface of the solid support (silica gel). If the distance between adjacent grafted particles is less than the size of a tracer molecule, it is not able to penetrate this space. Estimates made in [12] for Nautilus-E show that the average distance between adjacent selectors is indeed on the order of the diameter of a TtBB molecule.
Excess Adsorption Isotherms
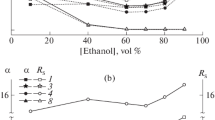
Excess adsorption isotherms of water from the binary mobile phase are shown in Fig. 1, in coordinates \(\Gamma _{1}^{{\left( n \right)}}\) and \(x_{1}^{l}\) (where indices 1 and 2 denote water and methanol, respectively). All isotherms are of the fifth type in Schay’s classification [17]; i.e., they have points of the adsorption azeotrope (\(\Gamma _{1}^{{\left( n \right)}} = 0\)) and characterize antibiotic CSPs as relatively hydrophilic (excessive adsorption of water is observed up to its mole fraction in mobile phases of 0.6–0.75 for H2О–МеОН and 0.8–0.9 for H2O–MeCN). The hydrophilicity of CSP grows in the order teicoplanin < ristocetin A < eremomycin.

Excess adsorption isotherms of water from (a) water–methanol and (b) water–acetonitrile mixtures on CSPs (1) Nautilus-E, (2) Chirobiotic R, and (3) Chirobiotic T.
The maximum excess adsorption of water from a mixture with acetonitrile is an order of magnitude higher than for a water–methanol solution. Two factors explain this behavior. The first of these is that it can compete with water molecules for interaction with proton-acceptor centers of the CSP surface, due to the proton-donor hydroxyl group in the methanol molecule. The second is associated with the structures of H2О–МеОН and H2O–MeCN. The first mixture is characterized by the exothermic effect of mixing and a relatively high degree of short-range order; the second, endothermic mixing and disordering (an increase in entropy) of the structure of a solution of relatively pure components [24, 25]. Contact between H2О–МеОН and a hydrophilic surface thus does not appreciably change the degree of ordering in the surface layer relative to the bulk phase, and increasing the concentration of water in it does not produce a notable gain in energy. In contrast, the accumulation of water at the interface in H2O–MeCN increases the degree of ordering and is thus energetically favorable, due to the drop in entropy. This also affects the thickness of the adsorption layer. As we can see from Table 2, the value of τ for an aqueous-acetonitrile solution is several times higher than for a mixture of water with methanol, since the ordering effect for H2O–MeCN acts on several molecular layers and gradually weakens. A similar effect was observed in [9] for another polar CSP, Whelk-O1.
The height of the H2O–MeOH surface layer is less than the size of the molecules of its components (0.3–0.4 nm). This is in obvious contradiction to the assumptions made in determining τ, the most important of which are the uniformity of the composition and the thickness of the adsorption phase over the adsorbent’s surface. Since the solvent fills the space between the grafted selectors, the thickness of the layers above and between a grafted particle will differ. It is also logical to assume that the molecules of the organic component preferably solvate hydrophobic ones, while the water molecules solvate hydrophilic regions of the selector and the free surface of the carrier. The composition of the adsorption phase will thus differ for different points on the surface, so τ is the apparent average thickness of the surface layer. The figures in Table 2 indicate that for a water–methanol mixture, its average thickness does not exceed one monomolecular layer.
Special attention should be given to the adsorption of water from nonaqueous organic solvents. The retained volume of a perturbation grows sharply for the water–acetonitrile mobile phase when the concentration of water is below 0.5 vol % (\(x_{1}^{l} < 0.014\)) (Fig. 2). Growth of function \({{V}_{R}}(x_{1}^{l})\) near a pure organic solvent is also observed for the water–methanol mobile phase, but it is much less than for H2O–MeCN. We believe a small fraction of strong Brønsted centers (possibly residual silanol groups of the support) lies on the surfaces of the adsorbents, which are deactivated by the small amount of water in the mobile phase. This effect is more pronounced for acetonitrile because methanol itself can to some extent deactivate these centers.

Dependences of the retained volume of a minor perturbation on the mole fraction of water in the mobile phase for (1) water–methanol and (2) water–acetonitrile mixtures.
Activity Coefficients in the Adsorption Phase
Figure 3 shows the dependences of the activity coefficients of the considered mixtures’ components on the mole fraction of water in the bulk phase. Similar dependences are shown in Fig. 4 for the activity coefficients of the components in the adsorption phase. Deviations from the properties of an ideal solution are observed for both binary liquids. These deviations are more pronounced in a system with the participation of acetonitrile. The latter is explained by acetonitrile (in contrast to methanol) not being part of the structures of short-range order formed by hydrogen bonds. Instead, it aggregates into clusters [25].

(a) Dependence of the activity coefficients of the components of (1) a water–methanol solution and (2) water. (b) Dependence of the activity coefficients of the components of (1) a water–acetonitrile solution and (2 ʹ) the mole fraction of water.

Dependences of the activity coefficients of the components of adsorption solutions of (a) water–methanol and (b) water–acetonitrile for CPSs (1) Nautilus-E, (2) Chirobiotic R, and (3) Chirobiotic T.
The behavior of the components of H2О–МеОН in the adsorption phase (Fig. 4a) is close to ideal (γa ≈ 1) in the range of a pure solvent (water (\(x_{1}^{a} = 1\)) or methanol (\(x_{1}^{a} = 0\))), up to the composition of the adsorption azeotrope. At the azeotropic point, there is a sharp, S-shaped drop in the activity coefficients down to values \(\gamma _{1}^{a}\) = 0.60–0.73 and \(\gamma _{2}^{a}\) = 0.43–0.56. These remain approximately constant in a certain range of compositions of the mobile phase, but continue to fall for water when \(x_{1}^{a} < 0.3\), and for methanol when \(x_{1}^{a} > 0.8\). Similar dependences γa on xa were observed in [26] for excess adsorption isotherms with azeotropic points. Values γa < 1 indicate additional stabilization of the substance in the adsorption phase, relative to the state of an ideal adsorption layer.
Deviations from the ideal for H2O–MeCN (Fig. 4b) are characterized by values γa > 1 (i.e., weaken adsorption relative to the state of an ideal adsorption layer). In contrast to methanol-containing mobile phases in the graphs of dependence γa on xa, there is no pronounced feature in the region of the adsorption azeotrope, and the form of these graphs in the range of \(x_{1}^{a} \in [0.2;0.98]\) resembles similar curves for the liquid phase, where the activity coefficients for water and acetonitrile grow monotonically as their concentrations fall (Fig. 3b). This is apparently associated with the height of the adsorption layer, which for H2O–MeCN is so high that its properties are to somewhat similar to those of a bulk liquid. Outside the specified range (i.e., near pure solvents), the entropy factor that ensures the thickness of the adsorption phase for acetonitrile solutions disappears. The adsorption phase grows thinner, and dependence γa(xa) is distorted. This behavior is not observed for water–methanol mixtures, since monomolecular adsorption layers form in them.
The imperfect behavior in the considered systems could be due to different violations of the model of an ideal adsorption layer, from the viewpoints of both the solid (the energy inhomogeneity of the surface) and the adsorption phase. The analysis performed in [12] showed that adsorbate–adsorbate interactions (rather than adsorbate–adsorbent interactions) are responsible for the shapes of the graphs in Fig. 4. The structuring of the adsorption solution on the CSPs surface seems to be the main reason for the observed patterns. For H2O–MeOH, this structuring means that the activity coefficients, which have values greater than 1 in the bulk liquid, are less than 1 in the adsorbed solution. With H2O–MeCN, we also observe a substantial (up to 3 times) drop in the values of γ upon moving from a bulk liquid to a surface solution of the same composition.
CONCLUSIONS
It was shown that CSPs with grafted macrocyclic antibiotics are hydrophilic materials on whose surfaces monomolecular adsorption layers form when in contact with H2О–МеОН. Polymolecular layers form when the CSPs are in contact with H2O–MeCN. The considered CSPs are characterized by qualitatively identical patterns of adsorption of aqueous-organic mixtures and differ slightly only in quantitative indicators of this adsorption.
REFERENCES
A. V. Kiselev, Intermolecular Interactions in Adsorption and Chromatography (Vyssh. Shkola, Moscow, 1986) [in Russian].
J. Mallette, M. Wang, and J. F. Parcher, Anal. Chem. 82, 3329 (2010). https://doi.org/10.1021/ac100148b
F. Gritti and G. Guiochon, Anal. Chem. 77, 4257 (2005). https://doi.org/10.1021/ac0580058
K. László, G. Nagy, G. Fóti, and G. Schay, Period. Polytech. Chem. Eng. 29 (2), 73 (1985).
Yu. V. Kazakevich, O. G. Larionov, V. O. Ulogov, and Yu. A. El’tekov, Zh. Fiz. Khim. 63, 3376 (1989).
Y. V. Kazakevich, R. LoBrutto, F. Chan, and T. Patel, J. Chromatogr., A 913, 75 (2001). https://doi.org/10.1016/s0021-9673(00)01239-5
F. Chan, L. S. Yeung, R. LoBrutto, and Y. V. Kazakevich, J. Chromatogr., A 1082, 158 (2005). https://doi.org/10.1016/j.chroma.2005.05.078
B. Buszewski, S. Bocian, and A. Felinger, J. Chromatogr., A 1191, 72 (2008). https://doi.org/10.1016/j.chroma.2007.11.074
L. Asnin, K. Horváth, and G. Guiochon, J. Chromatogr., A 1217, 1320 (2010). https://doi.org/10.1016/j.chroma.2009.12.066
A. Cavazzini, G. Nadalini, V. Malanchin, et al., Anal. Chem. 79, 3802 (2007). https://doi.org/10.1021/ac062240o
A. Cavazzini, G. Nadalini, V. Costa, and F. Dondi, J. Chromatogr., A 1143, 134 (2007). https://doi.org/10.1016/j.chroma.2006.12.090
Y. K. Nikitina, I. Ali, and L. D. Asnin, J. Chromatogr., A 1363, 71 (2014). https://doi.org/10.1016/j.chroma.2014.08.062
I. Poplewska, R. Kramarz, W. Piatkowski, et al., J. Chromatogr., A 1173, 58 (2007). https://doi.org/10.1016/j.chroma.2007.09.076
I. Poplewska, R. Kramarz, W. Piatkowski, et al., J. Chromatogr., A 1192, 130 (2008). https://doi.org/10.1016/j.chroma.2008.03.056
I. Ilisz, Z. Pataj, A. Aranyi, and A. Peter, Sep. Pur. Rev. 41, 207 (2012).https://doi.org/10.1080/15422119.2011.596253
E. A. Guggenheim and N. K. Adam, Proc. R. Soc. London, Ser. A 139, 218 (1933). https://doi.org/10.1098/rspa.1933.0015
G. Schay, L. G. Nagy, and T. Szekrenyesy, Period. Polytech. Chem. Eng. 4, 95 (1960).
O. G. Larionov and A. L. Myers, Chem. Eng. Sci. 26, 1025 (1971). https://doi.org/10.1016/0009-2509(71)80016-7
T. F. Anderson and J. M. Prausnitz, Ind. Eng. Chem. Process Des. Dev. 17, 552 (1978). https://doi.org/10.1021/i260068a028
A. Fredenslund, J. Gmehling, and P. Rasmussen, Vapor–Liquid Equilibria Using UNIFAC (Elsevier, Amsterdam, 1977).
A. Gordon and R. Ford, The Chemists Companion (Wiley, New York, 1972).
J. H. Knox and R. Kaliszan, J. Chromatogr. 349, 211 (1985). https://doi.org/10.1016/S0021-9673(01)83779-1
Y. V. Kazakevich and H. M. McNair, J. Chromatogr. Sci. 31, 317 (1993). https://doi.org/10.1093/chromsci/31.8.317
C. Moreau and G. Douheret, Thermochim. Acta 13, 385 (1975). https://doi.org/10.1016/0040-6031(75)85079-9
A. Wakisaka, H. Abdoul-Carime, Y. Yamamoto, and Y. Kiyozumi, J. Chem. Soc. Faraday Trans. 94, 369 (1998). https://doi.org/10.1039/A705777F
A. V. Kiselev and V. V. Khopina, Trans. Faraday Soc. 65, 1936 (1969). https://doi.org/10.1039/TF9696501936
Funding
This work was supported by the Russian Science Foundation, project no. 18-13-00240.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klimova, Y.A., Asnin, L.D. Adsorption of Binary Solvents on Chiral Stationary Phases with Grafted Macrocyclic Antibiotics. Russ. J. Phys. Chem. 95, 2304–2309 (2021). https://doi.org/10.1134/S0036024421110091
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036024421110091