
Abstract
Metabolism is a critical determinant of immune cell functionality. Immunometabolism, by definition, is a multidisciplinary area of immunology research that integrates the knowledge of energy transduction mechanisms and biochemical pathways. An important concept in the field is metabolic switch, a transition of immune cells upon activation to preferential utilization of select catabolic pathways for their energy needs. Mitochondria are not inert in this process and contribute to the metabolic adaptation by different mechanisms which include increasing ATP production to match dynamic bioenergetic demands and serving as a signaling platform. The latter involves generation of reactive oxygen species (ROS), one of the most intensively studied mitochondrial processes. While the role of mitochondrial ROS in the context of oxidative stress is well established, ROS signaling in immunity is an emerging and quickly changing field. In this review, we discuss ROS signaling and immunometabolism concepts from the standpoint of bioenergetics. We also provide a critical insight into the methodology for ROS assessment, outlining current challenges in the field. Finally, based on our analysis of the literature data, we hypothesize that regulatory ROS production, as opposed to oxidative stress, is controlled by mitochondrial biogenesis rather than metabolic switches.
Similar content being viewed by others
INTRODUCTION
During the past decades an entire literature dedicated to mitochondrial ROS (mitoROS) has been generated, signifying the importance of the topic in biology research. In 2002, we stated a simple kinetic model of ROS generation as a bimolecular reaction between oxygen and mitochondrial redox centers prone to auto-oxidation (ROS centers) [1]. According to the mass action law, the rate of such reaction is proportional to quantities (concentrations) of both reactants. The model also takes into account a dynamic nature of the ROS centers constantly cycling between reduced state capable of reacting with oxygen and a nonreactive oxidized state. ROS generation is therefore proportional both to the net concentration of ROS centers and to the fraction of their reduced form.
It is generally accepted that a major source of mitoROS production in the electron transport chain (ETC) is Complex I in its highly reduced state that can be produced by the specific Complex I inhibitor, rotenone, or other ETC inhibitors and conditions (such as cytochrome c depletion). Alternatively, rotenone can inhibit mitoROS production in a reverse electron transport (RET) scenario in hyperpolarized mitochondria [2-4]. This phenomenon is manifested under pathological conditions promoting excessive succinate accumulation and activation of proximal ROS producing sites. Notably, RET-induced oxidative stress was reported in ischemia-reperfusion injury [5-7]. However, as discussed below, the contribution of RET to ROS signaling seems unlikely under normal physiological conditions. Another canonical ROS center is Complex III, as was described in pioneering studies in 1972 [8, 9]. Complex III-derived ROS production is remarkably robust but typically manifests only in the presence of the specific inhibitor, antimycin A, that artificially elevates the level of unstable semiquinone form of coenzyme Q (an electron donor) on the matrix side of the Q-cycle. It is unclear whether such specific Complex III modification can be produced by natural effectors, and therefore, the role of this ROS center in physiological milieu remains debatable [2, 4, 10, 11]. An often-overlooked aspect of mitoROS production is their mitochondrial matrix sources as opposed to ETC. Previous studies suggested that mitochondrial matrix dehydrogenases contribute to ROS production in normally functioning mitochondria (in the absence of ETC inhibitors). In particular, a major ROS-producing activity is associated with dihydrolipoyl dehydrogenase (Dld) component of α-ketoglutarate and pyruvate dehydrogenase complexes [12, 13].
Historically, ROS production was associated with oxidative stress - by definition, a plethora of damaging effects to the cell. Evidence for regulatory roles of ROS currently lead to a paradigm shift towards “moderate” ROS as important signaling entities. Metabolic reprogramming of activated immune cells, specifically, T cells and macrophages, is associated with major bioenergetic changes. This is expected to affect both the mitochondrial mass/net number of ROS centers and their redox status, i.e., both components in the ROS generation model noted above [1]. These factors need to be considered to delineate ROS signaling pathways.
This review aims to provide critical insights into selected metabolic ROS-signaling mechanisms. We introduce key immunometabolism concepts, such as glycolytic switch and “broken” TCA cycle (tricarboxylic acid cycle), and discuss the role of metabolites with prominent immunomodulatory effects (“immunometabolites”). This constitutes the necessary background for a more in-depth analysis of bioenergetics of immune cells and their propensity to generate mitoROS. Further, we review proposed mechanisms of ROS signaling in immunity, in particular, the link between mitoROS and HIF1 function. Other highlights include mitoROS as damage-associated molecular patterns (DAMPs), the intrinsic mediators of proinflammatory responses. An interplay of mitoROS and non-mitoROS (primarily, NOX-derived H2O2 production in macrophages) is briefly discussed as well. The narrative is concluded with critical assessment of research tools extensively used in the field to ward off potential misinterpretation of experimental results. A broader bibliography on discussed topics can be found in specialized reviews referenced in each section.
The main conclusions drawn from the analysis of concepts outlined above are the following: (i) the regulatory mitoROS signaling, most likely, involves “mild” changes associated with mitochondrial biogenesis, as opposed to drastic changes in mitochondrial metabolic state typical for oxidative stress conditions. (ii) The metabolic changes in immune response generally favor increased energy transduction via the most effective mechanism, oxidative phosphorylation, as opposed to switch to glycolysis. (iii) Classically activated macrophages represent an exception due to their bactericidal function that requires massive production of reactive substances (NO and H2O2) that damage their own mitochondria.
IMMUNOMETABOLISM: GENERAL ASPECTS
An unprecedented interest in metabolic regulation of innate and adaptive immunity along with the development of multi-parameter metabolic flux analysis and high-precision “omic” profiling of immune cells shaped a new multidisciplinary subject of research, immunometabolism. A number of recent reviews have reintroduced fundamental metabolic pathways in the context of specialized immune cell functions and highlighted advances in the field (e.g. [14-19]). Here, we outline some of the recent immunometabolism concepts with an emphasis on glycolytic shift and concomitant changes in mitochondrial energy transduction.
Glycolytic switch concept. Reminiscent of the classical Warburg effect in cancer cells, normal immune cells (specifically, T cells and macrophages) rapidly shift from mitochondrial oxidative phosphorylation toward aerobic glycolysis when exposed to antigens, cytokines, and other stimuli. Although this metabolic change is well-documented (with early reports of increased glycolysis activity in stimulated lymphocytes dating back to the 60-70s [20, 21]), the obligatory requirement of aerobic glycolysis for immune cell activation and effector responses is still not completely understood. Since effector functions of immune cells, in particular cytokine production in lymphocytes, are generally associated with enhanced proliferative capacity [22], increased glycolytic capacity is thought to be necessary for various biosynthetic reactions supporting cell growth [14, 23-25]. Paradoxically, a hallmark of immune cell activation is increased lactate production that formally precludes utilization of upstream metabolites for de novo biosynthesis of macromolecules [15]. Moreover, a “switch” from the more efficient mitochondrial ATP production to aerobic glycolysis seems inconsistent with higher energy demand imposed by anabolic reactions, proliferation, and effector function per se.
Indeed, metabolic phenotypes of different types of activated immune cells (most notably, T cells and macrophages) and their functionally distinct subsets are not uniform. While glycolytic switch in activated T cells occurs concomitantly with rapid stimulation of mitochondrial respiration, stimulation of macrophages leads to iNOS upregulation and NO-mediated inhibition of ETC activity [26]. (This is further discussed in the section focused on the bioenergetics assessment of glycolytic switch).
Glycolytic enzymes control effector functions. Recent implications of glycolytic enzymes and metabolites in epigenetic and post-transcriptional control of specific effector functions suggest a more decisive role of glycolysis in immune response [27-30]. Specifically, interferon gamma (IFNγ) expression in T cells was shown to be negatively regulated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) binding to IFNγ mRNA; the translational constrain was relieved upon T cell activation-induced glycolytic shift and GAPDH engagement in energy production [27]. The inverse relationship between GAPDH availability and cytokine translation suggested an additional non-enzymatic role of GAPDH as a constitutive translation inhibitor in quiescent (non-activated) cells. More recently, GAPDH was implicated in an analogous mechanism of metabolic regulation of tumor necrosis factor α (TNFα) translation in macrophage pro-inflammatory response [31]. These studies are consistent with earlier work highlighting GAPDH propensity to bind AU-rich elements (AREs) of mRNA and alter protein expression [32]. It has been shown that AREs are displaced from binding to GAPDH by both substrates, glyceraldehyde 3-phosphate (G-3P) and NAD+ [33, 34]. Accordingly, G-3P potentiates production of IFNγ in glycolytically-deficient T-cells growing on slowly metabolized galactose but not in cells growing on glucose that already have higher levels of G-3P [27] (Fig. 1, a-c). It should be noted, however, that this is the scenario in which glycolytic flux is restricted upstream of GAPDH. If glycolytic flux is restricted downstream of GAPDH one should expect an exactly opposite effect upon lifting the restriction; the buildup of all intermediates, including G-3P, should cease. This is the hypothetical scenario of “metabolic switch” from oxidative phosphorylation (when glycolytic ATP-producing enzymes downstream of GAPDH are partially suppressed by mitochondrial competition) to glycolytic ATP production (Fig. 1d).

Mechanisms of regulation of ARE-mRNA abundance by glycolytic flux. Enzymes may be regulated by fluxes only indirectly through changes in metabolite abundances. Accordingly, the moonlighting mRNA-binding function of GAPDH is regulated by abundance of its substrates, G-3P and pyridine nucleotides. GAPDH, mRNA and a skeleton scheme of glycolytic pathway are shown in green, red and blue, respectively; basal lactate production is inconsequential and omitted for simplicity (a-c). Width of the arrows depicts the magnitude of the fluxes and the size of the font depicts the abundance of metabolites. (X) Indicates the restriction point in the pathway; under normal conditions (a) the restriction points are ATP-producing enzymes that are suppressed by competition from mitochondrial ATP production. Galactose shares most of classical glycolytic pathway but is slow to enter it (b and c). This leads to GAPDH binding, and depleting, of ARE-mRNAs, including IFNγ (b), the process reversible by exogenous G-3P (c) [27]. A hypothetical glycolytic switch scenario (d) could lead to a decreased IFNγ mRNA (and protein), in apparent discrepancy with observed IFNγ production. This discrepancy would require additional research to resolve.
Another unresolved issue with this mechanism is that both the reaction product NADH and its structural analog ATP are also effective in displacing the AREs from GAPDH in micromolar concentrations [27]. Since both pyridine and adenine nucleotides are present intracellularly at millimolar levels they should effectively compete off any AREs under all conditions. To resolve this paradox, it has been speculated that there might be compartments with much lower nucleotide levels, either in nucleus or parts of cytosol [27], but this presumption will require testing.
An alternative mechanism coordinating glycolytic shift and cytokine production involves lactate dehydrogenase A (LDHA), the enzyme catalyzing pyruvate conversion to lactate. Based on genetic LDHA depletion experiments, it was reported that under conditions of aerobic glycolysis, IFNγ expression is controlled by LDHA via acetyl-coA-dependent epigenetic mechanism (histone acetylation) but not by the GAPDH-linked translational regulation [28]. Manifestation of either mechanism might be profoundly influenced by other activities of these housekeeping proteins, in particular in loss-of-function paradigms. GAPDH has been predicted to be a rate-limiting glycolytic enzyme, in addition to the classical rate-limiting enzymes (hexokinase, phosphofructokinase, and pyruvate kinase) [35], while LDHA defines the end-point reaction of aerobic glycolysis. As such, both GAPDH and LDHA are important determinants of steady state levels of glycolytic metabolites, NAD+/NADH redox balance, and cell survival capacities. Explored as potential cancer therapy targeting Warburg metabolism, LDHA inhibition compromises serine and aspartate biosynthesis. However, in malignant cells, this effect is counteracted by activation of major pro-survival gene expression program mediated by ATF4, a transcription factor from cAMP-response element binding protein (CREB) family [36]. Reminiscent of this phenomenon, proliferation and effector function of activated Th1 cells involves ATF4-linked transcriptional response, in particular under conditions of amino acid deficiency [37]. Furthermore, lactate per se directly modulates immune cell functions [38-40] tending to decrease cytokine production at high extracellular concentrations. A recent study explored a novel lactate-driven epigenetic mechanism (histone lysine lactylation) linked to metabolic reprogramming in macrophages exposed to bacterial pathogens. Lactylation was increased by the mitochondrial inhibitor rotenone demonstrating a link between oxidative phosphorylation (mitochondrial) activity and transcriptional regulation. Indeed, lactate abundance inversely depends on mitochondrial pyruvate utilization that stops once pyridine nucleotide pool is fully reduced in the presence of rotenone [29]. LDHA-dependent epigenetic modulation of cytokine expression is also directly linked to mitochondrial metabolism, specifically citrate production in the Krebs cycle. Stimulated CD4+ T cells deficient in LDHA showed increased mitochondrial respiration and TCA cycle activity, while cytosolic acetyl-coA was decreased [28]. This suggested that, in wild type cells, glycolytic shift diverted citrate from intramitochondrial utilization and enhanced its export from mitochondria, thereby promoting cytosolic acetyl-coA generation from citrate, selective histone acetylation and IFNγ transcription [28]. Indeed, acetyl-CoA is also utilized for other cell functions, such as fatty-acid and cholesterol biosynthesis, and both histone and non-histone protein acetylation in various signaling pathways [41, 42].
With regard to citrate metabolism, mitochondrial citrate-malate antiporter (SLC25A1) emerged as a potentially important regulator of cytokine production (in particular in macrophages) and cellular NADP+/NADPH balance [43, 44]. These and other recent studies constitute a growing subtopic of immunometabolism focused on the role of TCA cycle metabolites (reviewed in [44]) that we briefly discuss below.
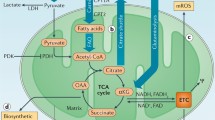
“Broken cycle” concept. Based on metabolic reprogramming patterns in macrophages and dendritic cells, it is postulated that under conditions of “glycolytic switch” and/or impaired mitochondrial respiration (e.g., due to nitric oxide [NO] production in activated macrophages), TCA cycle is no longer coupled to ETC activity and this leads to accumulation of immune-modulatory intermediates, most notably citrate and succinate [44, 45]. However, suppression of electron transfer through ETC via the mechanism of respiratory control or by direct inhibition, is expected to cause nearly complete reduction of NAD+ to NADH. This should terminate TCA cycle reactions due to deficit of NAD+, the substrate of four reactions, including pyruvate dehydrogenase at the entry to the cycle. In this scenario, neither citrate nor succinate could be produced (Fig. 2, a and b). To allow anabolic reactions through TCA cycle go forward, the excess of reducing equivalents would need to be transported from mitochondria via malate/aspartate shuttle (Fig. 2c) or otherwise utilized [46] whether these activities are sufficient for the release of TCA cycle function needs to be determined experimentally.
Immunometabolites: anabolic building blocks versus regulators. Metabolites can serve as precursors of building blocks (fatty acids and amino acids) for proliferation and protein biosynthesis, and as regulatory molecules. With regard to anabolic mechanisms, two considerations are important. First, it is unlikely that cells can forgo energy efficiency for a “greater good” of anabolic metabolism, as implies glycolytic switch concept. From the biochemical standpoint, immune response is primarily expenditure of vast amounts of energy, whether for protein synthesis (for proliferation and differentiation; antibody and cytokine production), action of cytoskeletal motors (for morphological changes) or ion pumps (for ionotropic signaling). Most cells, including immune cells, typically possess spare respiratory capacity, including excess of TCA capacity over basal energy needs, which can be used for biosyntheses without need to suppress bioenergetics. Second, cells in an organism normally receive most building blocks (amino acids, fatty acids, cholesterol, etc.) with the diet. Capacity of organism to augment that supply is quite limited. Ten amino acids out of twenty are “essential” and two more are synthesized from essential amino acids. All polyunsaturated fatty acids are either essential or synthesized from them. Ability to produce palmitate (from citrate) or serine and glycine (from 3-phosphoglycerate), typically considered in metabolic reprogramming scenarios, could serve at best a moderate correction of dietary imbalances (excluding liver and adipose tissue specialized in lipogenesis [47]). For cells in vitro, this also means that standard medium might have to be supplemented if some nutrients become limiting.

Suppression and rescue of TCA cycle biosynthetic activity by NAD+ availability. The biosynthetic activity is exemplified by citrate synthesis considered essential for lipid biosynthesis. Pyridine nucleotide reactions are shown with blue arrows, “3x” denotes 3 NADH produces in one turn of TCA cycle. Width of the arrows depicts the magnitude of the fluxes and the size of the font depicts the abundance of metabolites. (X) Indicates suppression of ETC activity. a) Normal citrate production in intact oxidative phosphorylation. b) Low citrate production under conditions of suppressed electron transport chain (ETC), including via respiratory control in glycolytic switch scenario. c) A hypothetical rescue of citrate synthesis by reverse malate-aspartate (Mal-Asp) shuttle activity, oxidation of NADH by oxaloacetate (OAA). The arrow indicating NADH to NAD+ conversion is linked to Mal-Asp shuttle symbol (as opposed to OAA to malate conversion) for figure clarity. All NADH (and/or NAD+) symbols represent a single uniform NADH (and/or NAD+) pool.
Regulatory mechanisms appear more impactful than anabolic. With regard to regulatory functions, as noted above, citrate and succinate participate in post-translational and post-transcriptional regulation. Changes in their levels (in particular, in the “broken” TCA cycle model) are substantial and sufficient to elicit significant effects. Citrate in TLR4-stimulated bone marrow derived macrophages (BMDM) rises about 2-fold [48] which may lead to a similar increase in cytosolic acetyl-CoA levels and the rates of histone acetylation. Additionally, acetyl-CoA can be converted to malonyl-CoA through the action of acetyl-CoA carboxylase, and malonyl-CoA levels do rise approximately 2.5-fold resulting in malonylation of multiple proteins including GAPDH [49]. Malonylation of lysine 213 (or its mimicking mutation to glutamic acid) boosts enzymatic activity roughly 3-fold and completely releases sequestered TNFα transcript [49]. Thus, both energy and effector functions are upregulated with a single modification originating from the citrate increase.
Succinate also increases more that 2-fold in TLR4-activated (Toll-like receptor 4) BMDMs [48]. It inhibits prolyl hydroxylase domain enzyme (PHD) with IC50 about 0.5 mM, well in the range of extracellular concentrations [50]. Consistent with structural similarities of the two metabolites, this inhibition occurs in competitive fashion and is counteracted by the enzyme substrate α-ketoglutatate [48]. Accordingly, PHD activity should depend on the ratio of these metabolites rather than their individual levels. In TLR4-stimulated RAW 264.7 macrophages α-ketoglutarate remains unchanged as succinate rises about 3-fold shifting the ratio towards inhibition [51]. PHD inhibition prevents destabilization of HIF1α (reviewed in Mitochondrial ROS and HIF1 regulation section below) and allows transcription of multiple genes including the pro-inflammatory cytokine IL-1β [48].
The total pool of TCA cycle metabolites (including succinate and citrate) increases in activated macrophages implying anaplerosis [48]. Stable isotope tracing demonstrates that the largest anaplerotic contributor is glutamine-to-glutamate- to-α-ketoglutatate pathway [48]. Succinate could also accumulate as a result of inhibition of succinate dehydrogenase (SDH), for example, by itaconate (reviewed below) [51, 52]. However, there appears to be no classic crossover point at SDH, as the downstream intermediates fumarate and malate are not depleted and may even increase [48] suggesting a modest role of SDH in succinate accumulation. Stable isotope tracing ruled out succinate production directly from itaconate [51].
Itaconate is arguably the archetypical immunometabolite (reviewed in [53]). Undetectable in resting macrophages, it exponentially rises to millimolar levels within a few hours upon TLR4 activation becoming the most abundant dicarboxylate in the cell [51, 54]. This kinetics is on par with such prominently responsive metabolites as prostaglandins [55] and NO [56]. It reflects activity of highly inducible (nearly 200-fold) enzyme cis-aconitate decarboxylase (CAD; also known as immunoresponsive gene 1, IRG1, protein), that converts TCA cycle intermediate aconitate to itaconate [54]. Among mammalian cells, this reaction apparently almost exclusive for macrophages [57]. It is not exactly clear what causes aconitate which is usually presumed to be tightly bound in the active center of aconitase, to dissociate and react with CAD.
The effects of itaconate are a subject of debate: whether it is pro-inflammatory due to SDH inhibition and accumulation of succinate [51] or anti-inflammatory [50, 58]. Favoring the latter, cytokines IL-1β, IL-6, IL-18, IL-12 (but not TNFα) are slightly upregulated (by 20-30%) in CAD-knockout cells suggesting existence of immune suppression by itaconate in wild type cells. However, surprisingly, hif-1α transcript is increased by order(s) of magnitude in the knockout cells resulting in the elevated HIF1α protein [52] which per se could account for the modest pro-inflammatory effect. Due to presence of reactive double bond conjugated with carboxylic group, itaconic acid can participate in a reaction of generalized Michael addition type with protein thiols. Posttranslational modification of Kelch-like ECH-associated protein 1 (KEAP1) via formation of the Michael adduct with downstream activation of anti-inflammatory factor Nrf2 (nuclear factor erythroid 2-related factor 2) was suggested as mechanism of itaconate action [58]. Overall, however, given the robust itaconate response upon stimulation, it is likely that we do not know yet the full potential of this metabolite.
Signal transduction notes. Much interest in the immunometabolism field has focused on post-activation dynamic changes in lipid metabolism, in particular, glycolysis-driven de novo fatty acid synthesis (FAS). The metabolic shift from fatty acid oxidation (FAO) to FAS is positively regulated by the mammalian target of rapamycin (mTOR)-dependent signal transduction pathways that is counterbalanced by AMP-activated kinase (AMPK) kinase activity fueling FAO [15, 59]. The serine/threonine kinase mTOR couples nutrient and oxygen availability with appropriate transcription programs. This regulatory network controls various aspects of innate and adaptive immunity, including macrophage polarization, T cell glycolytic reprogramming, cellular migration, regulatory T cell (Treg) function, and cytokine production in different types of immune cells [15, 59-62]. Among pleiotropic effects of mTOR is enhanced mitochondrial biogenesis and oxidative metabolism [62]. In addition to the oxygen sensor HIF-1α (discussed below), other essential transcription factors that control metabolic reprogramming include the Ca2+/calcineurin-dependent nuclear factor of activated T cells (NFAT) [63, 64] and c-Myc (MYC proto-oncogene), a major direct regulator of pro-survival gene expression [23, 45, 60, 65]. Nuclear factor-kappa B (NF-κB) is a major transcriptional activator of inflammatory responses, with a recently emerged role of a negative regulator of excessive inflammation [66]. This mechanism involves a complex interplay between selective elimination of defective mitochondria (mitophagy) and the NLRP3 inflammasome activation [67]. These signaling network components have been a subject of intense research and discussed in detail in signal transduction themed reviews (e.g. [62, 66, 68]).
BIOENERGETIC ASSESSMENT OF THE GLYCOLYTIC SWITCH
A recurring theme in immunometabolism is the concept of metabolic switch, an obligatory transition of cells to utilization of specific metabolic pathways to drive immune function. The particular case of glycolytic switch includes a set of notions, such as transition to aerobic glycolysis, ROS signaling and immunometabolite activity, that, as a whole or in parts, are applied to studies of various immune cells. In its entirety, such mechanism was proposed, for example, for SARS-CoV-2-infected (severe acute respiratory syndrome coronavirus 2) monocytes [69] as well as lipopolysaccharide-stimulated (LPS) macrophages [70]. In this section we aim to assess these notions from biochemist’s point of view. Our analysis is based largely on the dataset from a comprehensive study of the glycolytic switch in TLR4-activated macrophages [70]. This study deconvolved a network of metabolic changes accompanying pro-inflammatory macrophage polarization that eventually manifested in HIF1α stabilization and cytokine production.
The notion of glycolytic switch implies that glycolytic pathway is upregulated to produce ATP faster than oxidative phosphorylation thus suppressing the latter through substrate (ADP) deprivation. Accordingly, inherent to this mechanism is the higher ATP to ADP ratio (ATP/ADP) after the switch to glycolysis. Contrary to that, in TLR4-stimulated BMDMs, ATP/ADP drops by ~40%, corresponding to increase in ADP levels from 5 to 8% of the total pool. ADP rise should have resulted in activation of respiration, instead, respiration decreases by about ~40%. This result implies a completely different mechanism: an active suppression of oxidative phosphorylation rather than respiratory control by substrate availability. NO is the most likely factor responsible for this suppression [71]. It has been known since 1989 that NO is produced by activated macrophages and causes respiratory inhibition of target cells [72]. NO reversibly inhibits cytochrome c oxidase (respiratory complex IV) in a manner competitive to oxygen. NO is produced by inducible nitric oxide synthase (iNOS) expressed upon activation; macrophages in culture produce up to 1 µM NO, sufficient to cause their respiratory inhibition [73]. Prolonged exposure to NO can also cause an irreversible inhibition of respiratory complex I [74]. Additional targets may include aconitase 2 and pyruvate dehydrogenase inhibition of which may cause observed suppression of OCR (oxygen consumption rate) upstream of the respiratory chain [75]. The volatile nature of this compound may account for significant variability in respiratory inhibition observed in different studies.
Metabolic flux analysis platforms designed for simultaneous measurements of oxygen consumption and extracellular acidification rates (OCR and ECAR [extracellular acidification rate], respectively) combined with advanced data analysis [76] help to quantitatively re-evaluate the relative contribution of oxidative phosphorylation and glycolysis to overall bioenergetics capacity of immune cells. The study [70] presents sufficiently detailed data for this analysis including rarely reported proton production rate (PPR); raw ECAR read-out is insufficient. The analysis is based on stoichiometries of overall oxidative phosphorylation and glycolytic pathways. ATP output of oxidative phosphorylation has been revised downward from canonical 36 ATP molecules per glucose [77] to 30-32 (based on ATP/O ratios from [78]); accordingly, we use ATP/O2 = 5.45 [79]. The textbook stoichiometry of glycolysis is one ATP per lactic acid (ATP/H+ = 1) [77]. However, calculation of glycolytic ATP production based on raw proton production rate (PPR) gives only the upper limit estimate because of interference from mitochondria-produced CO2. We converted the reported proton production rate (PPR) and OCR values to ATP production rates. In the quiescent state 200,000 BMDMs produce ~1500 pmoles ATP per minute; this production drops upon TLR4 stimulation by about 40%, to ~900 pmol/min. The contribution of glycolysis increases from ~3% and ~17%, for quiescent and stimulated cells, respectively. These metabolic changes do not reach an extent of a “switch” that may be perceived as preferential reliance on glycolysis for energy.
Thus, paradoxically, mitochondrial function is impaired in the TLR4-stimulated macrophages and glycolysis remains a minor contributor that cannot keep up with energy demand. This results in decreased ATP production and lower ATP/ADP inevitably leading to turning off some ATP consumers which, in quiescent cells, are usually thought to be essential (housekeeping) processes necessary for cell survival. In a broader biological sense, however, survival may well be not an essential function for inflammatory macrophages.
Treatment with glycolysis inhibitor 2-deoxyglucose suppresses macrophage immune response (cytokine production) and rescues animals in the models of bacterial infection and/or hyperinflammation [48, 80, 81]. These results underscore a fine tuning of energy requirements for the immune response: it is preserved despite the existing energy deficiency but is strongly dependent upon the residual energy provision from glucose metabolism. The latter, obviously, is not limited to glycolysis but involves oxidative metabolism of glycolytically-produced pyruvate in mitochondria, a question is to what degree other mitochondrial substrates could substitute for it.
It should be also noted that, despite upregulated glycolysis, the suppression of oxidative phosphorylation does not occur in alternatively activated (in response to IL-4) macrophages. On contrary, they undergo PGC-1β-mediated (peroxisome proliferator-activated receptor gamma coactivator-1 beta) mitochondrial biogenesis resulting in increased capacity for oxidative metabolism [82, 83] corresponding to elevated energy demand [71, 84, 85]. Similar changes in bioenergetics occur in T cells in response to stimulation of T cell receptor (TCR) where both glycolysis and oxidative phosphorylation are upregulated [27, 86]. Thus, although activated glycolysis is a signature response of various immune cells to stimuli, glycolytic switch appears to have a limited scope.
RET and ROS. Another notion that needs to be reassessed is the ROS production mediated by reverse electron transport (RET). It is plausible that the glycolytic shift might result in activation of this mechanism if certain benchmarks were met. It is known from experiments with isolated mitochondria that this mechanism is exquisitely sensitive to any depolarization of mitochondrial membrane because it is driven by membrane potential (Δψ) “uphill”, against redox potential of the carriers [87, 88]. For this mechanism to function, mitochondria should be essentially in a non-phosphorylating, “resting” state. This is not the case for mitochondria in stimulated BMDMs that may preserve more than half of their normal ATP-synthetic activity (see above). Direct measurements show that although the signal of Δψ-sensitive probe tetramethylrhodamine methyl ester (TMRM) does go up upon TLR4 stimulation, it does not approach the level of actual State 4 attained with oligomycin [70]. Besides, this change is proportional to an increase in fluorescence of Δψ-insensitive mitochondrial probe MitoTracker Green indicating likelihood of the response to increased mitochondrial volume rather than Δψ. Also, as originally termed, RET is actually a transport of electrons from succinate to NAD+ and is manifested in highly reduced state of NAD+/NADH. However, in the activated BMDMs, the pyridine nucleotides remain 90% oxidized. Overall, in our opinion, stimulated BMDMs do not possess the benchmark conditions for RET and RET-mediated ROS production. In the inhibitor analysis of ROS, mitochondrial inhibitors that are capable of acutely blocking RET were added 3 hours before cell stimulation [70] leaving a room for a multitude of alternative chronic effects. Thus, although ROS signaling via HIF1α stabilization may still be the mechanism, the origin of the ROS remains obscure. The abovementioned increase in MitoTracker Green staining [70] consistent with increased mitochondrial mass, as well as syntheses of additional mitochondrial DNA in activated BMDMs [89], implies biogenesis with concomitant increase in mitochondrial redox carriers, including ROS-producing centers. This may be per se sufficient to increase overall ROS production, that can be augmented by slightly reduced milieu (change from ~8 to ~13% NADH reduction [70]), possibly due to a partial block in the respiratory chain.
Succinate and ROS generation. In classical experiments with isolated mitochondria succinate has to be provided to fuel both RET-mediated and Complex III-dependent ROS production. In intact cells, however, succinate is intrinsically present as a metabolite so, unless TCA cycle metabolites are severely depleted, it is not obvious why exogenous succinate would be required. Also, if TCA cycle is indeed depleted, any anaplerotic intermediate would suffice to replenish it, for example, provision of glutamine, precursor of α-ketoglutarate, is much easier achieved than delivery of succinate that cannot permeate plasma membrane. Succinate delivery from alkylated derivative (e.g., diethyl succinate) depends on its cleavage by intracellular nonspecific esterases of unknown nature and activity. Those activities are sufficient for a trapping of various probes and/or inhibitors. However, their capacity to cleave enough of a major consumable molecule as a TCA cycle metabolite to sustain normal flux has not been proven. In particular, the study discussed above [70] shows that diethyl succinate (referred to as simply succinate there) fails to increase intracellular succinate pool. However, it exerts a distinct pattern of biological effects: stimulation of TLR4-dependent pro-inflammatory signaling (IL-1β production) and suppression of anti-inflammatory signaling (IL-10 and IL-1RA) with no effect on TNFα. Surprisingly, diethyl ester of butyl malonate, a dicarboxylate transporter inhibitor, has the same biological signature, whereas it is known to inhibit of succinate oxidation in isolated mitochondria by preventing its access to SDH; this discrepancy indicates a potential off-target effect for both diethyl esters. Additionally, diethyl succinate per se barely caused any ROS production (~40% at 5 mM) compared to 4-fold increase from the TLR4 ligand LPS; unfortunately, a test of combination of the two effectors that gives the distinct biological signature mentioned above is not reported. Unlike diethyl succinate supplementation, LPS elevates cellular succinate levels (in the absence of exogenous sources) to the extent 3 times over the effect of SDH inhibitor malonate. This result argues that TLR4-dependent metabolic changes go beyond redistribution within a combined pool of TCA cycle intermediates but cause up to 10-fold increase of the pool (anaplerosis). Such an increase is entirely consistent with proposed increase in mitochondrial mass (see above).
ρ0 cells as a model of the glycolytic switch. A model of inducible respiration deficient (ρ0) cells generated via expression of dominant negative DNA polymerase γ, originally described by one of us [90], is used to mimic “metabolic switch” and probe downstream roles of mitochondria-originated signals (e.g., [91, 92]). These cells lack functional respiratory chain which undermines their ability to oxidize NADH produced in the TCA cycle and keep the cycle going. This may result in diminished biosynthesis of citrate, its product, cytosolic acetyl-CoA and, therefore, reduced histone acetylation. Several specific sites lose histone acetylation in ρ0 cells implying potential suppression of gene expression as the consequence [91]. Histone deacetylation is reversed by expression of an alternative respiratory chain consisting of rotenone-insensitive NADH-ubiquinone oxidoreductase, mitochondrial (NDI1) and AOX, a result interpreted as re-activation of the TCA cycle via NADH oxidation. Unexpectedly, NAD+/NADH ratio is unaffected by the ρ0 phenotype [91] suggesting some different mechanisms of histone deacetylation. NDI1 + AOX are not capable of generating ATP and, consistently, proliferation of ρ0 cells is not rescued by their expression [91] showing the critical importance of bioenergetic rather than biosynthetic function. Minor fraction (10-15%) of proliferative capacity can be rescued by normalization of Δψ in the ρ0 cells. Based on this and on concomitant response of CellROX indicator, ROS signaling via HIF1-mediated pathway was suggested [91]. However, regulation of ROS production by Δψ is mediated by hyper-reduced state of mitochondria; the latter can be detected as hyper-reduced NAD+/NADH, and it is not the case in the ρ0 cells [91]. As an alternative explanation, Δψ is necessary for protein import to mitochondria including components of biosynthetic machinery critical for proliferation and/or the major ROS-producing enzyme Dld [12, 13]. Overall, the ρ0 model emphasizes importance of bioenergetics for cell function while the role of the glycolyic switch remains questionable. The mechanism of ROS production is not clear, and its relevance, given ruined respiratory chain in this model, uncertain.
MITOCHONDRIAL ROS AS SIGNALING ENTITIES IN IMMUNITY
Growing literature is focused on specific regulatory roles of mitochondria-derived ROS as opposed to their well-documented pro-apoptotic and/or damaging effects promoted by mitochondrial dysfunction under various pathological conditions. MitoROS acting in a signaling mode have been implicated in autophagy, mitophagy, mitochondrial biogenesis, cell cycle progression, and other specific cell functions controlled by redox-sensitive signal transduction and transcription factors. Proteins that can be functionally modified by ROS are catalogued in other reviews. The list of ROS targets includes ATF4, NF-κB, mitogen activated protein kinases (MAPK): extracellular signal-regulated kinases (ERKs), p38 MAPKs, and the c-Jun N-terminal kinases (JNKs) [93]; protein tyrosine phosphatases, and p53 [10, 94-103]. However, the mechanisms of ROS generation and signaling in the context of immunometabolic regulation awaits further investigations and perhaps re-evaluation of existing concepts.
T cell highlights. Emerging body of literature suggests that mitoROS are potentially indispensable factors for T cell activation, effector functions and differentiation [42, 49, 96, 102, 104-108]. An earlier study of TCR-activated cells described acute effects of TCR-dependent ROS production on signal transduction pathways regulating T cell proliferation and survival [109]. Surprisingly, ROS signaling entities produced distinct outcomes: hydrogen peroxide promoted a proliferative signal via extracellular receptor kinase (ERK)-dependent pathway, while the effect of superoxide anion was consistent with activating a pro-death pathway (involving FAS-ligand expression). Other functional outcomes such NFAT activation and IL-2 expression were ROS-independent [109]. Although the site of ROS generation was not identified in this study, the involvement of mitoROS could not be excluded. Based on chemical and genetic inactivation of ETC components, other studies linked mitochondrial effects on T cell responses to ROS signaling originated from ETC [104, 110]. In particular, CD4+ T cells deficient in Rieske iron-sulfur protein, participant in the classical ROS-producing site at Complex III, exhibited impaired Ca2+-dependent NFAT-IL-2 pathway, but other redox-dependent signaling pathways (such as ERK activity) were not affected by the knockout. Rescue experiments with exogenously generated H2O2 supported a ROS-dependent factor in IL-2 induction. Interestingly, glycolysis was dispensable for T cell activation and IL-2 production in this paradigm [104]. In another report, IL-2 and IL-4 production in activated T cells was decreased by the antioxidant N-acetyl cysteine (NAC) and reduced in Complex I-deficient cells that led the authors to implicate Complex I as the source of regulatory ROS required for the effector function [110]. TCR-induced cytokine expression and H2O2 production were also inhibited by rotenone (at concentrations significantly exceeding those typically needed to achieve Complex I inhibition) that leaves an open question as to whether the ROS signal of interest originated from Complex I. A less invasive approach (with respect to ETC functional intactness) is the use of conventional and mitochondria-specific antioxidants. Although this approach alone is not sufficient to identify molecular mechanisms of ROS signaling (such as the site of ROS generation and their recipient targets), it provides the necessary proof-of-principle results to substantiate ROS involvement in a particular signaling pathway. In the “tool-box” section below we discuss some currently used ROS scavenging systems.
ROS as “SOS”. Pathogen-associated (PAMPs) and damage-asociated (DAMPs) molecular patterns are recognized by innate immune receptors as danger signals that trigger an acute immune response culminating in pathogen clearance and tissue repair and regeneration. The immune response to DAMP is often termed sterile inflammation as it is driven by intracellular entities, in the absence of foreign pathogens [111]. Impactful studies of danger signal recognition in macrophages identified pathways to activation of the NLRP3 (nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 protein) inflammasome, a molecular platform integrating pro-caspase I and pro-inflammatory cytokines processing upon stress signals [67, 89, 112-115]. Inflammasome-activating DAMPs include mitochondria-derived molecules, such as ROS, cardiolipin and mitochondrial DNA [67, 89, 113, 116]. Released mitochondrial DNA is also engaged in antiviral innate immunity by triggering the cGAS-STING signaling pathways and type I interferon responses [117-119]. This underscores yet another important role of mitochondria in immunity, beyond the ATP-producing task and classical apoptotic signaling via cytochrome c release [120, 121]. To release and/or display these “SOS” signals, mitochondria have to be selectively permeabilized or damaged in some way, but the conduits for DAMP release remain to be defined. Components of mitochondrial apoptotic pores have been discussed in the context of DNA and ROS release and consequent NLRP3 activation. Mitochondrial apoptotic permeabilization involves two well-known distinct mechanisms: the mitochondrial permeability transition pore (MPTP) and Bax/Bak-dependent mitochondrial outer membrane permeabilization (MOMP) pore [122]. The latter mechanism does not compromise the inner mitochondrial membrane integrity and is considered immunologically inert, although it has been shown that effector caspase activation downstream of Bax/Bak-induced MOMP promotes NLRP3-dependent IL-1β production [123]. The non-selective MPTP may be permissive to DNA transport [124, 125]. Translocation of mitochondrial DNA via MPTP was recently suggested in experiments with TDP-43-mediated inflammation; however, this suggestion was based largely on an effect of MPTP inhibitor cyclosporine A that was applied at a dose ~10 times higher than a specific dose [126]. An effect on an alternative target, calcineurin was also not ruled out. In addition, MPTP is generally associated with excessive mitochondrial dysfunction and would require control mechanisms. A recent study described a regulatory mechanism that involves protein tyrosine phosphatase-2 (SHP2) interaction with the adenine nucleotide translocase 1 (ANT1), a component of permeability transition pore [PTP] (based on the classical PTP model). It is suggested that SHP2-mediated inhibition of ANT1 keeps DAMP release in check and prevents excessive inflammasome activation [127]. Since the predominant species of reactive oxygen is freely permeating H2O2, it is reasonable to predict that mitoROS signal to inflammasome activation involves relatively subtle mitochondrial dysregulation, as opposed to the major “damage” such as the classical mitochondrial permeability transition. In this regard, previously described reversible and/or low-conductance states of MPTP [10, 128, 129] deserve further attention as they may be linked to ROS signal transmission from functionally intact mitochondria.
NLRP3 inflammasome activation involves mitoROS generated by Complex I and Complex III (but not Complex II, as expected) [113]. Interestingly, both ROS generation and the downstream inflammatory immune response were inhibited by down-regulation of the voltage-dependent anion channel (VDAC) potentially linking the ROS signal to metabolite transport [113]. MitoROS also convey damage signal through oxidation of mitochondrial DNA, as only oxidized DNA binds and activates NLRP3 [89, 114]. Another notable recent study identified a key role of parkin-dependent mitophagy in NF-κB-dependent control of excessive of NLRP3-inflammasome activation [67]. This and other studies [130] highlight the importance of mitochondrial quality control system that integrates mitophagy, mitochondrial dynamics, and biogenesis in the control of innate immune response and inflammation.
MITOCHONDRIAL ROS AND HIF1 REGULATION
Although transcriptional factor HIF1 is currently broadly implicated in a variety of cellular adaptive processes including immune response, the idea of ROS regulating the stability of its critical subunit HIF1α originates from the adaptation to physiological hypoxia (0.3-3% oxygen). It has been proposed that, under these conditions, HIF1α stabilization is critically dependent on mitoROS signal, therefore positioning mitochondria as the sensor of oxygen levels [131]. Under normoxic conditions, any expressed HIF1α is unstable as it is quickly hydroxylated at a critical proline residue by one of the family of α-ketoglutarate-dependent oxygenases called prolyl hydroxylase domain (PDH) proteins. The hydroxylated form has a three orders of magnitude heightened affinity to ubiquitin ligase complex in which von Hippel-Lindau tumor suppressor (pVHL) plays a role of recognition component. Thus, HIF1α is quickly ubiquitinilated and targeted for proteosomal degradation (see [132] for review). PDH proteins are perfect sensors for oxygen, as their KM is just above the solubility of atmospheric oxygen in aqueous phase (230-250 µM) [133] so their activity should respond nearly linearly to oxygen tensions in hypoxic range. Any alternative oxygen sensing mechanism should therefore imply that this fundamental, mass action control is somehow lost and the activity of the enzyme remains high despite virtual lack of one of the substrates, oxygen. Devising and proving such an intricate scenario may become a challenging enzymological task for future researchers.
An argument in favor of the mitochondrial ROS-mediated mechanism is increased responses of ROS-sensitive dyes under hypoxic conditions [92, 131, 134]. These data should be interpreted with some caution, especially when kinetic data are lacking [92, 134]. For example, an observed reversible change in fluorescence of dichlorofluorescin [131] may be difficult to explain. Responses of multiple mitochondrial ROS producing sites to various oxygen tensions have been studied in the less complex system, isolated mitochondria, with somewhat varying results but none of the studies detected elevated ROS production in the hypoxic range [135-137]. It was concluded that the ROS responses observed in cells are not intrinsic to mitochondria [136] and how they occur is unclear [95]. Exogenous hydrogen peroxide appears to stabilize HIF1α in a catalase-sensitive manner [92] lending some support to the idea that non-mitochondrial ROS may play a role in the process. However, the underlying question as to how do ROS inhibit HIF1α hydroxylation remains to be explored.
NON-MITOCHONDRIAL ROS IN IMMUNITY
The primary focus of this review is mitoROS, however, the topic of ROS signaling would be incomplete without touching upon other sources of ROS that may be even more potent. In response to pathogen invasion, phagocytic cells release enormous amounts of ROS in the process called respiratory burst. This process is executed by enzymes of the NADPH oxidase (NOX) family (the seven known members of the family are reviewed in [138]). The main player involved in immune response, NOX2 is a multimeric complex that assembles on the plasma membrane of the cell in a tightly regulated process in response to extracellular signals. It utilizes intracellular reducing equivalents in the form of NADPH to reduce oxygen with release of superoxide (and, eventually, its product hydrogen peroxide) into extracellular milieu [138, 139]. In the case of phagocytic cells, macrophages and neutrophils, these ROS play a major role in killing invading pathogens but may also signal back to the cell or to the other cells. For example, they suppress T cells responses to TCR activation in co-cultures of neutrophils and T cells [140]. In the absence of phagocytes, endogenous NOX-derived ROS may also play a role in activation-induced cell death in previously activated T cells (reviewed in [106]). In addition to these immunosuppressive mechanisms, there are reports of ROS involvement in activation of T cells. TCR activation causes extracellular superoxide generation, apparently via NOX2, but surprisingly it is not required for T cell activation, proliferation and cytokine production [141]. Based on these and other data, it was concluded that NOX2 is required for proper differentiation to Th2, and away from Th17, phenotype but not the activation of T cells. Analysis of effects of antioxidants led to a hypothesis that lipid-soluble peroxidation products, or their enzymatically derived counterparts, leukotrienes, rather than ROS per se are the acting entities in T cell activation [106]. NOX-generated ROS have been implicated in NLRP3 inflammasome activation in macrophages (reviewed in [142]). There are several lines of evidence supporting this notion. Whereas specificity of NOX inhibitors can be questioned [143], complexity of NOX responses, involving translocation and assembly of multiple subunits, paradoxically lend support to the model through genetic manipulations and localization studies (reviewed in [142]). Activation of inflammasome in response to particulate matter also fits in the model of phagocytosis-driven NOX activation and ROS production, analogous to the respiratory burst [144]. Thus, the NOX enzymes appear to be a viable alternative to mitochondria as the source of ROS signaling. There is no apparent reason why a ROS signal coming from NOX would be any different from a one originated in mitochondria, as in both cases it would most likely be the same relatively stable and freely permeating H2O2. The relative contributions of the two sources were recently determined in cells not specializing in phagocytosis, myoblasts, for which there is no apparent reason to expect extra high activity of NOXs. However, their contribution was substantial, around 40% [145]. A similar analysis in different types of immune cells would significantly enhance our understanding of the immune ROS signaling.
Reciprocally, as NOXs contribute to signaling, mitochondria may contribute to oxidative burst and bactericidal activity. This has been reported for both macrophages [146] and neutrophils [147]. Importantly, in the process mitochondria are actively recruited to the sites of bactericidal activity in NOX2-containing phagosomes [146]. A positive feedback mechanism between mitochondrial and NOX-dependent ROS production may occur leading to amplification of bactericidal activity [147-149].
TOOLBOX CONTENT
As the results of any study are only as good as the methods employed, we will briefly touch upon some key tools used in the studies of ROS and metabolic signaling.
ROS probes. Some of the methods to assess production rates and resulting levels of ROS along with potential missteps and pitfalls in their application were discussed in our previous review on the subject of ROS with emphasis on dichlorofluorescin and its esterified analog (H2-DCF-DA or DCF-DA) [11]. In recent years, however, a positively charged tri-phenyl-phosphonium derivative of dihydroethydium (DHE), MitoSOX, gained popularity in the ROS research as a supposedly specific sensor of intramitochondrial superoxide. It should be noted however, that it is a subject to all limitations of Δψ-sensitive fluorescent indicators [150, 151]. MitoSOX fluorescence depends not only on ROS reactivity but also on reversible compartmentalization between mitochondrial matrix, cytosol and external medium that is determined by Δψ on both mitochondrial and plasma membranes; any changes in Δψ will change probe levels and fluorescence in matrix and cytosol. Another potential variable could be differential binding to mitochondrial versus nuclear DNA [151]. Besides, the probe uncouples mitochondria and inhibits cytochrome c oxidase (Complex IV) at very low concentrations [152] making control for those parameters obligatory for meaningful interpretation of the data. Existence of more than one fluorescent oxidation product makes this probe purely qualitative unless measured with HPLC [153]. Although there are no very reliable probes for ROS, MitoSOX appears to be particularly difficult for use.
Antioxidants. NAC. Although broadly employed as a generalized antioxidant it should be rather considered as a reducing agent and moderate sulfhydryl protector; it can also play a role in glutathione replenishment but only under conditions of severe depletion [154, 155]. The free radical scavenging mechanism was strongly questioned based on the low-rate constants of reaction with superoxide [156-158]. NAC is weakly reactive with H2O2 and superoxide [157]. Reaction of NAC with superoxide is a chain reaction in which superoxide is regenerated; the chain reaction terminates with conversion of superoxide to another ROS, H2O2 [157]. Metabolism to produce more reductive low molecular weight sulfur compounds has been described [158] but its relevance to ROS detoxification requires further validation.
SkQ and MitoQ. Mitochondria-targeted quinones MitoQ and, to a lesser degree, its close structural analog SkQ [159, 160] also became widely used to probe ROS signaling with implied specificity for mitochondrially generated ROS [159, 161]. It should be noted, however, that the spatial selectivity of these compounds was inferred based of proximity and compartmentalization considerations [161]. On the other hand, SkQ and MitoQ are effective scavengers of free radicals in membranes, including the ones that are generated from the exogenous peroxide [162]. Given that the bulk of cellular ROS is the freely diffusible hydrogen peroxide, protection afforded by mitochondrially targeted quinones may be a questionable predictor of the ROS origin. Both compounds were developed with an idea of catalytic antioxidant in mind [159, 160] as both are structural analogs of a natural redox carrier coenzyme Q which is undergoing redox cycling in the respiratory chain. The MitoQ is a close coenzyme Q mimic, whereas structural features of SkQ were optimized for best reactivity and membrane permeation [160, 161]. The ability of SkQ to be reduced by complexes I and II, and therefore regenerate, was directly demonstrated [160]. It should be noted that both compounds are actually strong prooxidants at micromolar concentrations [160], so special care should be taken to not overdose. The submicromolar concentrations do show antioxidant properties with SkQ demonstrating broader therapeutic window than MitoQ [160]. Overall, the SkQ appears to have advantages over MitoQ and, in our opinion, should be a compound of choice in studies of oxidative stress and ROS signaling. On the other hand, any conclusions about the source of ROS based on its effects would be speculative at best.
Ester forms of metabolites. Since many of immunometabolites are plasma membrane impermeable, use of alkylated derivatives became a matter of such a routine in the studies of immunometabolism that even the name of original metabolite is used in place of its derivative [70, 163]. It should not be overlooked, however, that those derivatives are distinct compounds with their unique chemistries rather than mere precursors of desired metabolites. For example, di-methyl itaconate is much more reactive than itaconate in formation of Michael adducts with proteins [58] as is another esterified dicarboxylic acid derivative dimethyl fumarate in comparison to fumarate [164]. On the other hand, di-methyl itaconate is not metabolized to itaconate inside the cells, so its use would completely defeat the purpose [165]. To circumvent this, 4-octyl itaconate was proposed as an alternative [58]. However, it gives completely discordant effects versus endogenous itaconate on particulate matter-induced inflammatory response in BMDM. The authors noted important differences between endogenous itaconate and the exogenously-applied octly ester [163]. There is no detectable increase in the desired metabolite succinate in the BMDM supplemented with dimethyl succinate [70]. Remarkably, the use of the esters may be even unnecessary in the specific case of macrophages. It is possible to increase intracellular itaconate to millimolar range with just prolonged incubation with unmodified compound [51]. It is also seems possible to effectively deliver canonical membrane-impermeant molecules, such as coenzyme A and carboxyatractlyoside [84], apparently owing to the phagocytic nature of macrophages. Obviously, other strategies may be required for T cells and other immune cells. Thus, it is advisable to monitor changes in the desired metabolite levels upon application of a derivative and control for the effects of the cleaved groups (converted to methanol or ethanol for methyl and ethyl derivatives, respectively). A useful strategy would be to use control non-hydrolysable analogs (as is done to distinguish between enzymatic and non-enzymatic effects of ATP).
CONCLUDING REMARKS
The conceptual distinction between ROS signaling and oxidative stress is primarily quantitative: “moderate” ROS produce regulatory signals, while excessive ROS overwhelm ROS defense systems and damage cell constituents. Somewhere in between, ROS may not be the direct damaging agents, but initiate a regulated pathway for cell self-destruction in the form of apoptosis.
The steep dependence of ROS production on redox status of ETC carriers provides a direct link to the metabolic state of the cells. This underlying notion prompted us to extend this review of ROS signaling-related data to include bioenergetic aspects of immunometabolism. It appears that the harmless mode of ROS signaling that safeguards against oxidative stress is the basal ROS production. It is augmented, upon stimulation, by mitochondrial biogenesis rather than activation through a sharp metabolic switch. Consistently, bioenergetic function remains sustained or upregulated for energy-demanding functions, like proliferation. However, for macrophages, the sentinel cells, upon discovery of an invader, production of cytotoxic molecules (NO and H2O2) apparently takes precedence over energy efficiency resulting in self-injury and suppressed bioenergetics.
In our analysis, we highlighted some controversial mechanisms (such as RET) not to downplay the data but rather to evoke further interest in the matter. The area of research, at crossroads of immunometabolism and ROS signals, is full of unresolved mysteries and stunning paradoxes. It presents an unplowed field for researchers willing to devote themselves to resolving finest details of immensely complex and extremely important mechanisms of immunity.
Abbreviations
- ANT1:
-
adenine nucleotide translocase 1
- ARE:
-
AU-rich element
- AOX:
-
alternative oxidase, mitochondrial
- ATF4:
-
cyclic AMP-dependent transcription factor ATF-4
- BMDM:
-
bone marrow derived macrophages
- CAD:
-
cis-aconitate decarboxylase, EC:4.1.1.6
- DAMP:
-
damage-asociated molecular pattern
- ERK:
-
extracellular receptor kinase
- ETC:
-
electron transport chain
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase, EC:1.2.1.12
- G-3P:
-
glyceraldehyde 3-phosphate
- HIF1:
-
hypoxia-induced factor 1
- HIF1α:
-
alpha subunit of hypoxia-induced factor 1
- IFNγ:
-
interferon gamma
- IL:
-
interleukin
- LDHA:
-
lactate dehydrogenase A, EC:1.1.1.27
- MitoQ:
-
10-(6-ubiquinoyl) decyltriphenyl-phosphonium
- mitoROS:
-
mitochondria-produced ROS
- mTOR:
-
mammalian target of rapamycin
- Nrf2:
-
nuclear factor erythroid 2-related factor 2
- NAC:
-
N-acetyl cysteine
- NFAT:
-
nuclear factor of activated T cells
- NLRP3:
-
nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3 protein
- NO:
-
nitric oxide
- NOX:
-
NADPH oxidase
- OCR:
-
oxygen consumption rate
- PHD:
-
prolyl hydroxylase domain enzyme
- RET:
-
reverse electron transport
- ROS:
-
reactive oxygen species
- SDH:
-
succinate dehydrogenase
- SkQ:
-
10-(6′-plastoquinonyl) decyltriphenylphosphonium
- TCA cycle:
-
tricarboxylic acid cycle
- TCR:
-
T cell receptor
- TLR4:
-
Toll-like receptor 4, EC:3.2.2.6
- TMRM:
-
tetramethylrhodamine methyl ester
- TNFα:
-
tumor necrosis factor α
- Δψ:
-
membrane potential
- ρ0 :
-
respiration deficient cells
References
Kushnareva, Y., Murphy, A. N., and Andreyev, A. (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state, Biochem. J., 368, 545-553, https://doi.org/10.1042/BJ20021121.
Andreyev, A. Y., Kushnareva, Y. E., and Starkov, A. A. (2005) Mitochondrial metabolism of reactive oxygen species, Biochemistry (Moscow), 70, 200-214, https://doi.org/10.1007/s10541-005-0102-7.
Scialo, F., Fernandez-Ayala, D. J., and Sanz, A. (2017) Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease, Front. Physiol., 8, 428, https://doi.org/10.3389/fphys.2017.00428.
Brand, M. D. (2010) The sites and topology of mitochondrial superoxide production, Exp. Gerontol., 45, 466-472, https://doi.org/10.1016/j.exger.2010.01.003.
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijevic, D., Sundier, S. Y., et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS, Nature, 515, 431-435, https://doi.org/10.1038/nature13909.
Niatsetskaya, Z. V., Sosunov, S. A., Matsiukevich, D., Utkina-Sosunova, I. V., Ratner, V. I., Starkov, A. A., and Ten, V. S. (2012) The oxygen free radicals originating from mitochondrial complex I contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice, J. Neurosci., 32, 3235-3244, https://doi.org/10.1523/JNEUROSCI.6303-11.2012.
Sahni, P. V., Zhang, J., Sosunov, S., Galkin, A., Niatsetskaya, Z., Starkov, A., Brookes, P. S., and Ten, V. S. (2018) Krebs cycle metabolites and preferential succinate oxidation following neonatal hypoxic-ischemic brain injury in mice, Pediatr. Res., 83, 491-497, https://doi.org/10.1038/pr.2017.277.
Boveris, A., and Chance, B. (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen, Biochem. J., 134, 707-716, https://doi.org/10.1042/bj1340707.
Boveris, A., Oshino, N., and Chance, B. (1972) The cellular production of hydrogen peroxide, Biochem. J., 128, 617-630, https://doi.org/10.1042/bj1280617.
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release, Physiol. Rev., 94, 909-950, https://doi.org/10.1152/physrev.00026.2013.
Andreyev, A. Y., Kushnareva, Y. E., Murphy, A. N., and Starkov, A. A. (2015) Mitochondrial ROS metabolism: 10 years later, Biochemistry (Moscow), 80, 517-531, https://doi.org/10.1134/S0006297915050028.
Starkov, A. A., Fiskum, G., Chinopoulos, C., Lorenzo, B. J., Browne, S. E., Patel, M. S., and Beal, M. F. (2004) Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species, J. Neurosci., 24, 7779-7788, https://doi.org/10.1523/JNEUROSCI.1899-04.2004.
Kareyeva, A. V., Grivennikova, V. G., Cecchini, G., and Vinogradov, A. D. (2011) Molecular identification of the enzyme responsible for the mitochondrial NADH-supported ammonium-dependent hydrogen peroxide production, FEBS Lett., 585, 385-389, https://doi.org/10.1016/j.febslet.2010.12.019.
Buck, M. D., O’Sullivan, D., and Pearce, E. L. (2015) T cell metabolism drives immunity, J. Exp. Med., 212, 1345-1360, https://doi.org/10.1084/jem.20151159.
Almeida, L., Lochner, M., Berod, L., and Sparwasser, T. (2016) Metabolic pathways in T cell activation and lineage differentiation, Semin. Immunol., 28, 514-524, https://doi.org/10.1016/j.smim.2016.10.009.
Diskin, C., and Palsson-McDermott, E. M. (2018) Metabolic modulation in macrophage effector function, Front. Immunol., 9, 270, https://doi.org/10.3389/fimmu.2018.00270.
O’Neill, L. A., Kishton, R. J., and Rathmell, J. (2016) A guide to immunometabolism for immunologists, Nat. Rev. Immunol., 16, 553-565, https://doi.org/10.1038/nri.2016.70.
Ramalho, R., Rao, M., Zhang, C., Agrati, C., Ippolito, G., Wang, F. S., Zumla, A., and Maeurer, M. (2020) Immunometabolism: new insights and lessons from antigen-directed cellular immune responses, Semin. Immunopathol., 42, 279-313, https://doi.org/10.1007/s00281-020-00798-w.
Escoll, P., and Buchrieser, C. (2018) Metabolic reprogramming of host cells upon bacterial infection: why shift to a Warburg-like metabolism? FEBS J., 285, 2146-2160, https://doi.org/10.1111/febs.14446.
Wang, T., Marquardt, C., and Foker, J. (1976) Aerobic glycolysis during lymphocyte proliferation, Nature, 261, 702-705, https://doi.org/10.1038/261702a0.
Bennett, W. E., and Cohn, Z. A. (1966) The isolation and selected properties of blood monocytes, J. Exp. Med., 123, 145-160, https://doi.org/10.1084/jem.123.1.145.
Gudmundsdottir, H., Wells, A. D., and Turka, L. A. (1999) Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity, J. Immunol., 162, 5212-5223.
Delmastro-Greenwood, M. M., and Piganelli, J. D. (2013) Changing the energy of an immune response, Am. J. Clin. Exp. Immunol., 2, 30-54.
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation, Science, 324, 1029-1033, https://doi.org/10.1126/science.1160809.
Hume, D. A., Radik, J. L., Ferber, E., and Weidemann, M. J. (1978) Aerobic glycolysis and lymphocyte transformation, Biochem. J., 174, 703-709, https://doi.org/10.1042/bj1740703.
Wang, A., Luan, H. H., and Medzhitov, R. (2019) An evolutionary perspective on immunometabolism, Science, 363, https://doi.org/10.1126/science.aar3932.
Chang, C. H., Curtis, J. D., Maggi, L. B., Jr., Faubert, B., Villarino, A. V., et al. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis, Cell, 153, 1239-1251, https://doi.org/10.1016/j.cell.2013.05.016.
Peng, M., Yin, N., Chhangawala, S., Xu, K., Leslie, C. S., and Li, M. O. (2016) Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism, Science, 354, 481-484, https://doi.org/10.1126/science.aaf6284.
Zhang, D., Tang, Z., Huang, H., Zhou, G., Cui, C., et al. (2019) Metabolic regulation of gene expression by histone lactylation, Nature, 574, 575-580, https://doi.org/10.1038/s41586-019-1678-1.
Phan, A. T., Goldrath, A. W., and Glass, C. K. (2017) Metabolic and epigenetic coordination of T cell and macrophage immunity, Immunity, 46, 714-729, https://doi.org/10.1016/j.immuni.2017.04.016.
Millet, P., Vachharajani, V., McPhail, L., Yoza, B., and McCall, C. E. (2016) GAPDH binding to TNF-alpha mRNA contributes to posttranscriptional repression in monocytes: a novel mechanism of communication between inflammation and metabolism, J. Immunol., 196, 2541-2551, https://doi.org/10.4049/jimmunol.1501345.
Colell, A., Green, D. R., and Ricci, J. E. (2009) Novel roles for GAPDH in cell death and carcinogenesis, Cell Death Differ., 16, 1573-1581, https://doi.org/10.1038/cdd.2009.137.
Nagy, E., and Rigby, W. F. (1995) Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD+-binding region (Rossmann fold), J. Biol. Chem., 270, 2755-2763, https://doi.org/10.1074/jbc.270.6.2755.
Rodriguez-Pascual, F., Redondo-Horcajo, M., Magan-Marchal, N., Lagares, D., Martinez-Ruiz, A., Kleinert, H., and Lamas, S. (2008) Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability, Mol. Cell. Biol., 28, 7139-7155, https://doi.org/10.1128/MCB.01145-08.
Liberti, M. V., Dai, Z., Wardell, S. E., Baccile, J. A., Liu, X., et al. (2017) A predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product, Cell Metab., 26, 648-659 e648, https://doi.org/10.1016/j.cmet.2017.08.017.
Pathria, G., Scott, D. A., Feng, Y., Sang Lee, J., Fujita, Y., et al. (2018) Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival, EMBO J., 37, https://doi.org/10.15252/embj.201899735.
Yang, X., Xia, R., Yue, C., Zhai, W., Du, W., et al. (2018) ATF4 regulates CD4+ T cell immune responses through metabolic reprogramming, Cell Rep., 23, 1754-1766, https://doi.org/10.1016/j.celrep.2018.04.032.
Dietl, K., Renner, K., Dettmer, K., Timischl, B., Eberhart, K., et al. (2010) Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes, J. Immunol., 184, 1200-1209, https://doi.org/10.4049/jimmunol.0902584.
Haas, R., Cucchi, D., Smith, J., Pucino, V., Macdougall, C. E., and Mauro, C. (2016) Intermediates of metabolism: from bystanders to signalling molecules, Trends Biochem. Sci., 41, 460-471, https://doi.org/10.1016/j.tibs.2016.02.003.
Haas, R., Smith, J., Rocher-Ros, V., Nadkarni, S., Montero-Melendez, T., et al. (2015) Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions, PLoS Biol., 13, e1002202, https://doi.org/10.1371/journal.pbio.1002202.
Metallo, C. M., and Vander Heiden, M. G. (2010) Metabolism strikes back: metabolic flux regulates cell signaling, Genes Dev., 24, 2717-2722, https://doi.org/10.1101/gad.2010510.
Weinberg, S. E., Sena, L. A., and Chandel, N. S. (2015) Mitochondria in the regulation of innate and adaptive immunity, Immunity, 42, 406-417, https://doi.org/10.1016/j.immuni.2015.02.002.
Palmieri, E. M., Spera, I., Menga, A., Infantino, V., Porcelli, V., et al. (2015) Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation, Biochim. Biophys. Acta, 1847, 729-738, https://doi.org/10.1016/j.bbabio.2015.04.009.
Williams, N. C., and O’Neill, L. A. J. (2018) A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation, Front. Immunol., 9, 141, https://doi.org/10.3389/fimmu.2018.00141.
Loftus, R. M., and Finlay, D. K. (2016) Immunometabolism: cellular metabolism turns immune regulator, J. Biol. Chem., 291, 1-10, https://doi.org/10.1074/jbc.R115.693903.
Chinopoulos, C. (2020) Acute sources of mitochondrial NAD+ during respiratory chain dysfunction, Exp. Neurol., 327, 113218, https://doi.org/10.1016/j.expneurol.2020.113218.
Hillgartner, F. B., Salati, L. M., and Goodridge, A. G. (1995) Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis, Physiol. Rev., 75, 47-76, https://doi.org/10.1152/physrev.1995.75.1.47.
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-McDermott, E. M., McGettrick, A. F., et al. (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha, Nature, 496, 238-242, https://doi.org/10.1038/nature11986.
Gill, T., and Levine, A. D. (2013) Mitochondria-derived hydrogen peroxide selectively enhances T cell receptor-initiated signal transduction, J. Biol. Chem., 288, 26246-26255, https://doi.org/10.1074/jbc.M113.476895.
Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., et al. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase, Cancer Cell, 7, 77-85, https://doi.org/10.1016/j.ccr.2004.11.022.
Cordes, T., Wallace, M., Michelucci, A., Divakaruni, A. S., Sapcariu, S. C., et al. (2016) Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels, J. Biol. Chem., 291, 14274-14284, https://doi.org/10.1074/jbc.M115.685792.
Lampropoulou, V., Sergushichev, A., Bambouskova, M., Nair, S., Vincent, E. E., et al. (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation, Cell Metab., 24, 158-166, https://doi.org/10.1016/j.cmet.2016.06.004.
O'Neill, L. A. J., and Artyomov, M. N. (2019) Itaconate: the poster child of metabolic reprogramming in macrophage function, Nat. Rev. Immunol., 19, 273-281, https://doi.org/10.1038/s41577-019-0128-5.
Michelucci, A., Cordes, T., Ghelfi, J., Pailot, A., Reiling, N., et al. (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production, Proc. Natl. Acad. Sci. USA, 110, 7820-7825, https://doi.org/10.1073/pnas.1218599110.
Dennis, E. A., Deems, R. A., Harkewicz, R., Quehenberger, O., Brown, H. A., et al. (2010) A mouse macrophage lipidome, J. Biol. Chem., 285, 39976-39985, https://doi.org/10.1074/jbc.M110.182915.
Salkowski, C. A., Detore, G., McNally, R., van Rooijen, N., and Vogel, S. N. (1997) Regulation of inducible nitric oxide synthase messenger RNA expression and nitric oxide production by lipopolysaccharide in vivo: the roles of macrophages, endogenous IFN-gamma, and TNF receptor-1-mediated signaling, J. Immunol., 158, 905-912.
Strelko, C. L., Lu, W., Dufort, F. J., Seyfried, T. N., Chiles, T. C., Rabinowitz, J. D., and Roberts, M. F. (2011) Itaconic acid is a mammalian metabolite induced during macrophage activation, J. Am. Chem. Soc., 133, 16386-16389, https://doi.org/10.1021/ja2070889.
Mills, E. L., Ryan, D. G., Prag, H. A., Dikovskaya, D., Menon, D., et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1, Nature, 556, 113-117, https://doi.org/10.1038/nature25986.
Newton, R., Priyadharshini, B., and Turka, L. A. (2016) Immunometabolism of regulatory T cells, Nat. Immunol., 17, 618-625, https://doi.org/10.1038/ni.3466.
Lindsten, T., June, C. H., and Thompson, C. B. (1988) Multiple mechanisms regulate c-myc gene expression during normal T cell activation, EMBO J., 7, 2787-2794.
Weichhart, T., Hengstschlager, M., and Linke, M. (2015) Regulation of innate immune cell function by mTOR, Nat. Rev. Immunol., 15, 599-614, https://doi.org/10.1038/nri3901.
Saravia, J., Raynor, J. L., Chapman, N. M., Lim, S. A., and Chi, H. (2020) Signaling networks in immunometabolism, Cell Res., 30, 328-342, https://doi.org/10.1038/s41422-020-0301-1.
Muller, M. R., and Rao, A. (2010) NFAT, immunity and cancer: a transcription factor comes of age, Nat. Rev. Immunol., 10, 645-656, https://doi.org/10.1038/nri2818.
Seo, H., Chen, J., Gonzalez-Avalos, E., Samaniego-Castruita, D., Das, A., et al. (2019) TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion, Proc. Natl. Acad. Sci. USA, 116, 12410-12415, https://doi.org/10.1073/pnas.1905675116.
Wang, R., Dillon, C. P., Shi, L. Z., Milasta, S., Carter, R., et al. (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation, Immunity, 35, 871-882, https://doi.org/10.1016/j.immuni.2011.09.021.
Afonina, I. S., Zhong, Z., Karin, M., and Beyaert, R. (2017) Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome, Nat. Immunol., 18, 861-869, https://doi.org/10.1038/ni.3772.
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., et al. (2016) NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria, Cell, 164, 896-910, https://doi.org/10.1016/j.cell.2015.12.057.
Jones, R. G., and Pearce, E. J. (2017) MenTORing immunity: mTOR signaling in the development and function of tissue-resident immune cells, Immunity, 46, 730-742, https://doi.org/10.1016/j.immuni.2017.04.028.
Codo, A. C., Davanzo, G. G., Monteiro, L. B., de Souza, G. F., Muraro, S. P., et al. (2020) Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/glycolysis-dependent axis, Cell Metab., 32, 437-446 e435, https://doi.org/10.1016/j.cmet.2020.07.007.
Mills, E. L., Kelly, B., Logan, A., Costa, A. S. H., Varma, M., et al. (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages, Cell, 167, 457-470 e413, https://doi.org/10.1016/j.cell.2016.08.064.
Van den Bossche, J., Baardman, J., Otto, N. A., van der Velden, S., Neele, A. E., et al. (2016) Mitochondrial dysfunction prevents repolarization of inflammatory macrophages, Cell Rep., 17, 684-696, https://doi.org/10.1016/j.celrep.2016.09.008.
Stuehr, D. J., and Nathan, C. F. (1989) Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells, J. Exp. Med., 169, 1543-1555, https://doi.org/10.1084/jem.169.5.1543.
Brown, G. C., and Borutaite, V. (1999) Nitric oxide, cytochrome c and mitochondria, Biochem. Soc. Symp., 66, 17-25, https://doi.org/10.1042/bss0660017.
Clementi, E., Brown, G. C., Feelisch, M., and Moncada, S. (1998) Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione, Proc. Natl. Acad. Sci. USA, 95, 7631-7636, https://doi.org/10.1073/pnas.95.13.7631.
Palmieri, E. M., Gonzalez-Cotto, M., Baseler, W. A., Davies, L. C., Ghesquiere, B., et al. (2020) Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase, Nat. Commun., 11, 698, https://doi.org/10.1038/s41467-020-14433-7.
Mookerjee, S. A., Gerencser, A. A., Nicholls, D. G., and Brand, M. D. (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements, J. Biol. Chem., 292, 7189-7207, https://doi.org/10.1074/jbc.M116.774471.
Nicholls, D. G., Ferguson, S. J. (2001) Bioenergetics 3, Academic Press, San-Diego.
Lehninger, A. L. (1976) Biochemistry, 2nd edition, Worth Publishers Inc., New York, N. Y.
Divakaruni, A. S., Wallace, M., Buren, C., Martyniuk, K., Andreyev, A. Y., et al. (2017) Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death, J. Cell Biol., 216, 1091-1105, https://doi.org/10.1083/jcb.201612067.
Wang, A., Huen, S. C., Luan, H. H., Yu, S., Zhang, C., Gallezot, J. D., Booth, C. J., and Medzhitov, R. (2016) Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation, Cell, 166, 1512-1525.e1512, https://doi.org/10.1016/j.cell.2016.07.026.
Wang, A., Pope, S. D., Weinstein, J. S., Yu, S., Zhang, C., Booth, C. J., and Medzhitov, R. (2019) Specific sequences of infectious challenge lead to secondary hemophagocytic lymphohistiocytosis-like disease in mice, Proc. Natl. Acad. Sci. USA, 116, 2200-2209, https://doi.org/10.1073/pnas.1820704116.
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., et al. (2006) Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation, Cell Metab., 4, 13-24, https://doi.org/10.1016/j.cmet.2006.05.011.
Huang, S. C., Everts, B., Ivanova, Y., O’Sullivan, D., Nascimento, M., et al. (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages, Nat. Immunol., 15, 846-855, https://doi.org/10.1038/ni.2956.
Divakaruni, A. S., Hsieh, W. Y., Minarrieta, L., Duong, T. N., Kim, K. K. O., et al. (2018) Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis, Cell Metab., 28, 490-503.e497, https://doi.org/10.1016/j.cmet.2018.06.001.
Tan, Z., Xie, N., Cui, H., Moellering, D. R., Abraham, E., Thannickal, V. J., and Liu, G. (2015) Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism, J. Immunol., 194, 6082-6089, https://doi.org/10.4049/jimmunol.1402469.
Faris, R., Fan, Y. Y., De Angulo, A., Chapkin, R. S., deGraffenried, L. A., and Jolly, C. A. (2014) Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential for murine CD4+ T cell metabolic activation, Biochim. Biophys. Acta, 1842, 1475-1482, https://doi.org/10.1016/j.bbalip.2014.07.009.
Loschen, G., Flohe, L., and Chance, B. (1971) Respiratory chain linked H2O2 production in pigeon heart mitochondria, FEBS Lett., 18, 261-264, https://doi.org/10.1016/0014-5793(71)80459-3.
Korshunov, S. S., Skulachev, V. P., and Starkov, A. A. (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria, FEBS Lett., 416, 15-18, https://doi.org/10.1016/s0014-5793(97)01159-9.
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., et al. (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation, Nature, 560, 198-203, https://doi.org/10.1038/s41586-018-0372-z.
Jazayeri, M., Andreyev, A., Will, Y., Ward, M., Anderson, C. M., and Clevenger, W. (2003) Inducible expression of a dominant negative DNA polymerase-gamma depletes mitochondrial DNA and produces a rho0 phenotype, J. Biol. Chem., 278, 9823-9830, https://doi.org/10.1074/jbc.m211730200.
Martinez-Reyes, I., Diebold, L. P., Kong, H., Schieber, M., Huang, H., et al. (2016) TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions, Mol. Cell, 61, 199-209, https://doi.org/10.1016/j.molcel.2015.12.002.
Bell, E. L., Klimova, T. A., Eisenbart, J., Moraes, C. T., Murphy, M. P., Budinger, G. R., and Chandel, N. S. (2007) The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production, J. Cell Biol., 177, 1029-1036, https://doi.org/10.1083/jcb.200609074.
Son, Y., Kim, S., Chung, H. T., and Pae, H. O. (2013) Reactive oxygen species in the activation of MAP kinases, Methods Enzymol., 528, 27-48, https://doi.org/10.1016/B978-0-12-405881-1.00002-1.
Dard, L., Blanchard, W., Hubert, C., Lacombe, D., and Rossignol, R. (2020) Mitochondrial functions and rare diseases, Mol. Aspects Med., 71, 100842, https://doi.org/10.1016/j.mam.2019.100842.
Sena, L. A., and Chandel, N. S. (2012) Physiological roles of mitochondrial reactive oxygen species, Mol. Cell, 48, 158-167, https://doi.org/10.1016/j.molcel.2012.09.025.
Franchina, D. G., Dostert, C., and Brenner, D. (2018) Reactive oxygen species: involvement in T cell signaling and metabolism, Trends Immunol., 39, 489-502, https://doi.org/10.1016/j.it.2018.01.005.
Flohe, L., Brigelius-Flohe, R., Saliou, C., Traber, M. G., and Packer, L. (1997) Redox regulation of NF-kappa B activation, Free Radic. Biol. Med., 22, 1115-1126, https://doi.org/10.1016/s0891-5849(96)00501-1.
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017) NF-kappaB signaling in inflammation, Signal Transduct. Target. Ther., 2, https://doi.org/10.1038/sigtrans.2017.23.
Son, Y., Cheong, Y. K., Kim, N. H., Chung, H. T., Kang, D. G., and Pae, H. O. (2011) Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J. Signal Transduct., 2011, 792639, https://doi.org/10.1155/2011/792639.
Chandel, N. S., Vander Heiden, M. G., Thompson, C. B., and Schumacker, P. T. (2000) Redox regulation of p53 during hypoxia, Oncogene, 19, 3840-3848, https://doi.org/10.1038/sj.onc.1203727.
Liu, B., Chen, Y., and St Clair, D. K. (2008) ROS and p53: a versatile partnership, Free Radic. Biol. Med., 44, 1529-1535, https://doi.org/10.1016/j.freeradbiomed.2008.01.011.
Nathan, C., and Cunningham-Bussel, A. (2013) Beyond oxidative stress: an immunologist's guide to reactive oxygen species, Nat. Rev. Immunol., 13, 349-361, https://doi.org/10.1038/nri3423.
Zhang, H., Wang, L., and Chu, Y. (2019) Reactive oxygen species: the signal regulator of B cell, Free Radic. Biol. Med., 142, 16-22, https://doi.org/10.1016/j.freeradbiomed.2019.06.004.
Sena, L. A., Li, S., Jairaman, A., Prakriya, M., Ezponda, T., et al. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling, Immunity, 38, 225-236, https://doi.org/10.1016/j.immuni.2012.10.020.
Rashida Gnanaprakasam, J. N., Wu, R., and Wang, R. (2018) Metabolic reprogramming in modulating T cell reactive oxygen species generation and antioxidant capacity, Front. Immunol., 9, 1075, https://doi.org/10.3389/fimmu.2018.01075.
Belikov, A. V., Schraven, B., and Simeoni, L. (2015) T cells and reactive oxygen species, J. Biomed. Sci., 22, 85, https://doi.org/10.1186/s12929-015-0194-3.
Previte, D. M., O’Connor, E. C., Novak, E. A., Martins, C. P., Mollen, K. P., and Piganelli, J. D. (2017) Reactive oxygen species are required for driving efficient and sustained aerobic glycolysis during CD4+ T cell activation, PLoS One, 12, e0175549, https://doi.org/10.1371/journal.pone.0175549.
Reth, M. (2002) Hydrogen peroxide as second messenger in lymphocyte activation, Nat. Immunol., 3, 1129-1134, https://doi.org/10.1038/ni1202-1129.
Devadas, S., Zaritskaya, L., Rhee, S. G., Oberley, L., and Williams, M. S. (2002) Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression, J. Exp. Med., 195, 59-70, https://doi.org/10.1084/jem.20010659.
Kaminski, M. M., Sauer, S. W., Klemke, C. D., Suss, D., Okun, J. G., Krammer, P. H., and Gulow, K. (2010) Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression, J. Immunol., 184, 4827-4841, https://doi.org/10.4049/jimmunol.0901662.
Gong, T., Liu, L., Jiang, W., and Zhou, R. (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases, Nat. Rev. Immunol., 20, 95-112, https://doi.org/10.1038/s41577-019-0215-7.
Meylan, E., Tschopp, J., and Karin, M. (2006) Intracellular pattern recognition receptors in the host response, Nature, 442, 39-44, https://doi.org/10.1038/nature04946.
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011) A role for mitochondria in NLRP3 inflammasome activation, Nature, 469, 221-225, https://doi.org/10.1038/nature09663.
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis, Immunity, 36, 401-414, https://doi.org/10.1016/j.immuni.2012.01.009.
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T. M., et al. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome, Nat. Immunol., 12, 222-230, https://doi.org/10.1038/ni.1980.
Banoth, B., and Cassel, S. L. (2018) Mitochondria in innate immune signaling, Transl. Res., 202, 52-68, https://doi.org/10.1016/j.trsl.2018.07.014.
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., et al. (2015) Mitochondrial DNA stress primes the antiviral innate immune response, Nature, 520, 553-557, https://doi.org/10.1038/nature14156.
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., et al. (2014) Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production, Cell, 159, 1549-1562, https://doi.org/10.1016/j.cell.2014.11.036.
Rongvaux, A., Jackson, R., Harman, C. C., Li, T., West, A. P., et al. (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA, Cell, 159, 1563-1577, https://doi.org/10.1016/j.cell.2014.11.037.
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c, Cell, 86, 147-157, https://doi.org/10.1016/s0092-8674(00)80085-9.
Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and Newmeyer, D. D. (1997) The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis, Science, 275, 1132-1136, https://doi.org/10.1126/science.275.5303.1132.
Kushnareva, Y., and Newmeyer, D. D. (2010) Bioenergetics and cell death, Ann. N. Y. Acad. Sci., 1201, 50-57, https://doi.org/10.1111/j.1749-6632.2010.05633.x.
Vince, J. E., De Nardo, D., Gao, W., Vince, A. J., Hall, C., et al. (2018) The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1beta activation, Cell Rep., 25, 2339-2353.e2334, https://doi.org/10.1016/j.celrep.2018.10.103.
Zorov, D. B. (1996) Mitochondrial damage as a source of diseases and aging: a strategy of how to fight these, Biochim. Biophys. Acta, 1275, 10-15, https://doi.org/10.1016/0005-2728(96)00042-4.
Patrushev, M., Kasymov, V., Patrusheva, V., Ushakova, T., Gogvadze, V., and Gaziev, A. (2004) Mitochondrial permeability transition triggers the release of mtDNA fragments, Cell. Mol. Life Sci., 61, 3100-3103, https://doi.org/10.1007/s00018-004-4424-1.
Yu, C. H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., et al. (2020) TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS, Cell, 183, 636-649 e618, https://doi.org/10.1016/j.cell.2020.09.020.
Guo, W., Liu, W., Chen, Z., Gu, Y., Peng, S., et al. (2017) Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis, Nat. Commun., 8, 2168, https://doi.org/10.1038/s41467-017-02351-0.
Kushnareva, Y. E., and Sokolove, P. M. (2000) Prooxidants open both the mitochondrial permeability transition pore and a low-conductance channel in the inner mitochondrial membrane, Arch. Biochem. Biophys., 376, 377-388, https://doi.org/10.1006/abbi.2000.1730.
Ichas, F., Jouaville, L. S., and Mazat, J. P. (1997) Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals, Cell, 89, 1145-1153, https://doi.org/10.1016/s0092-8674(00)80301-3.
Xu, Y., Shen, J., and Ran, Z. (2020) Emerging views of mitophagy in immunity and autoimmune diseases, Autophagy, 16, 3-17, https://doi.org/10.1080/15548627.2019.1603547.
Chandel, N. S., Maltepe, E., Goldwasser, E., Mathieu, C. E., Simon, M. C., and Schumacker, P. T. (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription, Proc. Natl. Acad. Sci. USA, 95, 11715-11720, https://doi.org/10.1073/pnas.95.20.11715.
Schofield, C. J., and Ratcliffe, P. J. (2004) Oxygen sensing by HIF hydroxylases, Nat. Rev. Mol. Cell Biol., 5, 343-354, https://doi.org/10.1038/nrm1366.
Hirsila, M., Koivunen, P., Gunzler, V., Kivirikko, K. I., and Myllyharju, J. (2003) Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor, J. Biol. Chem., 278, 30772-30780, https://doi.org/10.1074/jbc.M304982200.
Bell, E. L., Klimova, T. A., Eisenbart, J., Schumacker, P. T., and Chandel, N. S. (2007) Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia, Mol. Cell. Biol., 27, 5737-5745, https://doi.org/10.1128/MCB.02265-06.
Oshino, N., Jamieson, D., and Chance, B. (1975) The properties of hydrogen peroxide production under hyperoxic and hypoxic conditions of perfused rat liver, Biochem. J., 146, 53-65, https://doi.org/10.1042/bj1460053.
Hoffman, D. L., Salter, J. D., and Brookes, P. S. (2007) Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling, Am. J. Physiol. Heart Circ. Physiol., 292, H101-H108, https://doi.org/10.1152/ajpheart.00699.2006.
Stepanova, A., Konrad, C., Manfredi, G., Springett, R., Ten, V., and Galkin, A. (2019) The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A, J. Neurochem., 148, 731-745, https://doi.org/10.1111/jnc.14654.
Panday, A., Sahoo, M. K., Osorio, D., and Batra, S. (2015) NADPH oxidases: an overview from structure to innate immunity-associated pathologies, Cell. Mol. Immunol., 12, 5-23, https://doi.org/10.1038/cmi.2014.89.
Brown, D. I., and Griendling, K. K. (2009) Nox proteins in signal transduction, Free Radic. Biol. Med., 47, 1239-1253, https://doi.org/10.1016/j.freeradbiomed.2009.07.023.
Cemerski, S., Cantagrel, A., Van Meerwijk, J. P., and Romagnoli, P. (2002) Reactive oxygen species differentially affect T cell receptor-signaling pathways, J. Biol. Chem., 277, 19585-19593, https://doi.org/10.1074/jbc.M111451200.
Belikov, A. V., Schraven, B., and Simeoni, L. (2014) TCR-triggered extracellular superoxide production is not required for T-cell activation, Cell Commun. Signal., 12, 50, https://doi.org/10.1186/s12964-014-0050-1.
Martinon, F. (2010) Signaling by ROS drives inflammasome activation, Eur. J. Immunol., 40, 616-619, https://doi.org/10.1002/eji.200940168.
Aldieri, E., Riganti, C., Polimeni, M., Gazzano, E., Lussiana, C., Campia, I., and Ghigo, D. (2008) Classical inhibitors of NOX NAD(P)H oxidases are not specific, Curr. Drug Metab., 9, 686-696, https://doi.org/10.2174/138920008786049285.
Dostert, C., Petrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., and Tschopp, J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica, Science, 320, 674-677, https://doi.org/10.1126/science.1156995.
Wong, H. S., Benoit, B., and Brand, M. D. (2019) Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts, Free Radic. Biol. Med., 130, 140-150, https://doi.org/10.1016/j.freeradbiomed.2018.10.448.
West, A. P., Brodsky, I. E., Rahner, C., Woo, D. K., Erdjument-Bromage, H., et al. (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS, Nature, 472, 476-480, https://doi.org/10.1038/nature09973.
Vorobjeva, N., Prikhodko, A., Galkin, I., Pletjushkina, O., Zinovkin, R., Sud’ina, G., Chernyak, B., and Pinegin, B. (2017) Mitochondrial reactive oxygen species are involved in chemoattractant-induced oxidative burst and degranulation of human neutrophils in vitro, Eur. J. Cell Biol., 96, 254-265, https://doi.org/10.1016/j.ejcb.2017.03.003.
Dikalov, S. (2011) Cross talk between mitochondria and NADPH oxidases, Free Radic. Biol. Med., 51, 1289-1301, https://doi.org/10.1016/j.freeradbiomed.2011.06.033.
Pinegin, B., Vorobjeva, N., Pashenkov, M., and Chernyak, B. (2018) The role of mitochondrial ROS in antibacterial immunity, J. Cell. Physiol., 233, 3745-3754, https://doi.org/10.1002/jcp.26117.
Nicholls, D. G. (2012) Fluorescence measurement of mitochondrial membrane potential changes in cultured cells, Methods Mol. Biol., 810, 119-133, https://doi.org/10.1007/978-1-61779-382-0_8.
Polster, B. M., Nicholls, D. G., Ge, S. X., and Roelofs, B. A. (2014) Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species, Methods Enzymol., 547, 225-250, https://doi.org/10.1016/B978-0-12-801415-8.00013-8.
Roelofs, B. A., Ge, S. X., Studlack, P. E., and Polster, B. M. (2015) Low micromolar concentrations of the superoxide probe MitoSOX uncouple neural mitochondria and inhibit complex IV, Free Radic. Biol. Med., 86, 250-258, https://doi.org/10.1016/j.freeradbiomed.2015.05.032.
Zielonka, J., and Kalyanaraman, B. (2010) Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth, Free Radic. Biol. Med., 48, 983-1001, https://doi.org/10.1016/j.freeradbiomed.2010.01.028.
Parasassi, T., Brunelli, R., Costa, G., De Spirito, M., Krasnowska, E., Lundeberg, T., Pittaluga, E., and Ursini, F. (2010) Thiol redox transitions in cell signaling: a lesson from N-acetylcysteine, ScientificWorldJournal, 10, 1192-1202, https://doi.org/10.1100/tsw.2010.104.
Samuni, Y., Goldstein, S., Dean, O. M., and Berk, M. (2013) The chemistry and biological activities of N-acetylcysteine, Biochim. Biophys. Acta, 1830, 4117-4129, https://doi.org/10.1016/j.bbagen.2013.04.016.
Benrahmoune, M., Therond, P., and Abedinzadeh, Z. (2000) The reaction of superoxide radical with N-acetylcysteine, Free Radic. Biol. Med., 29, 775-782, https://doi.org/10.1016/s0891-5849(00)00380-4.
Winterbourn, C. C., and Metodiewa, D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide, Free Radic. Biol. Med., 27, 322-328, https://doi.org/10.1016/s0891-5849(99)00051-9.
Ezerina, D., Takano, Y., Hanaoka, K., Urano, Y., and Dick, T. P. (2018) N-Acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production, Cell Chem. Biol., 25, 447-459.e444, https://doi.org/10.1016/j.chembiol.2018.01.011.
Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., Smith, R. A., and Murphy, M. P. (2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties, J. Biol. Chem., 276, 4588-4596, https://doi.org/10.1074/jbc.M009093200.
Skulachev, V. P. (2007) A biochemical approach to the problem of aging: “megaproject” on membrane-penetrating ions. The first results and prospects, Biochemistry (Moscow), 72, 1385-1396, https://doi.org/10.1134/s0006297907120139.
Skulachev, V. P. (2013) Cationic antioxidants as a powerful tool against mitochondrial oxidative stress, Biochem. Biophys. Res. Commun., 441, 275-279, https://doi.org/10.1016/j.bbrc.2013.10.063.
Antonenko, Y. N., Roginsky, V. A., Pashkovskaya, A. A., Rokitskaya, T. I., Kotova, E. A., Zaspa, A. A., Chernyak, B. V., and Skulachev, V. P. (2008) Protective effects of mitochondria-targeted antioxidant SkQ in aqueous and lipid membrane environments, J. Membr. Biol., 222, 141-149, https://doi.org/10.1007/s00232-008-9108-6.
Sun, K. A., Li, Y., Meliton, A. Y., Woods, P. S., Kimmig, L. M., Cetin-Atalay, R., Hamanaka, R. B., and Mutlu, G. M. (2020) Endogenous itaconate is not required for particulate matter-induced NRF2 expression or inflammatory response, Elife, 9, https://doi.org/10.7554/eLife.54877.
Brennan, M. S., Matos, M. F., Li, B., Hronowski, X., Gao, B., et al. (2015) Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro, PLoS One, 10, e0120254.
ElAzzouny, M., Tom, C. T., Evans, C. R., Olson, L. L., Tanga, M. J., Gallagher, K. A., Martin, B. R., and Burant, C. F. (2017) Dimethyl itaconate is not metabolized into itaconate intracellularly, J. Biol. Chem., 292, 4766-4769, https://doi.org/10.1074/jbc.C117.775270.
Funding
This work was supported in part by a grant from the National Institute of Health (P01AG014930/AG/NIA NIH HSS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. This article does not contain description of studies with the involvement of humans or animal subjects.
Rights and permissions
About this article

Cite this article
Andreyev, A.Y., Kushnareva, Y.E., Starkova, N.N. et al. Metabolic ROS Signaling: To Immunity and Beyond. Biochemistry Moscow 85, 1650–1667 (2020). https://doi.org/10.1134/S0006297920120160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920120160